
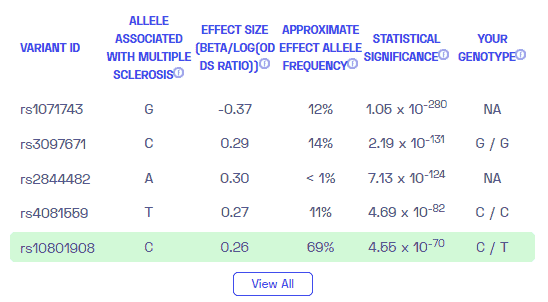
Informe de ADN de Nebula Genomics para EM
¿La EM es genética? Creamos un informe de ADN basado en un estudio que intentó responder esta pregunta. A continuación puede ver un informe de MUESTRA de ADN. Para obtener su informe de ADN personalizado, compre nuestro Secuenciación del genoma completo !

Información Adicional
¿Qué es la EM? (¿La parte 1 de la EM es genética?)
La esclerosis múltiple (EM) o encefalomielitis diseminada (DE) es una enfermedad autoinmune neurológica inflamatoria crónica con formas de progresión muy diferentes, según el área de daño nervioso, la extensión y la gravedad de la enfermedad. Específicamente, ataca las vainas de mielina en el cerebro y la médula espinal del sistema nervioso central (SNC). Las vainas de mielina son capas aislantes que protegen los nervios. Los ataques hacen que la vaina de mielina se inflame en pequeños parches (placas o lesiones), que se pueden ver en una resonancia magnética. La interrupción de los mensajes enviados a lo largo de los nervios provoca síntomas de EM.
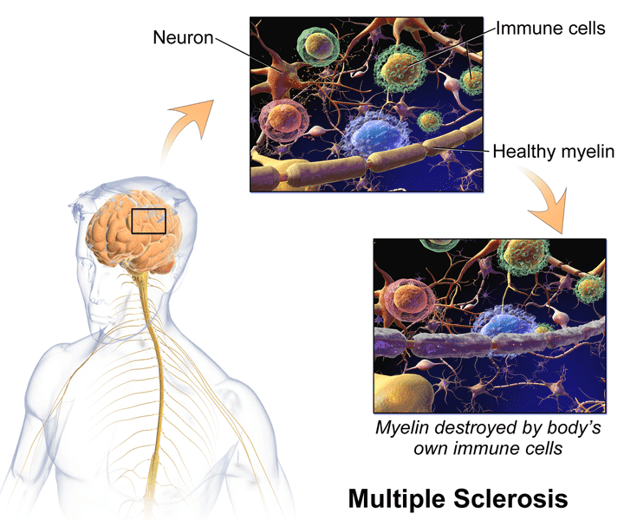
Hay dos tipos principales de EM: EM primaria progresiva, en la que la enfermedad se caracteriza por un empeoramiento de las funciones neurológicas sin recaídas ni remisiones, y EM remitente-recaída, en la que los síntomas tienden a presentarse en episodios distinguibles o recaídas. La EM secundaria progresiva sigue un curso inicial remitente-recurrente.
Las causas exactas de la enfermedad aún no se han aclarado a pesar de los esfuerzos de investigación. En la EM, muchos focos de desmielinización inflamatoria (múltiples) se encuentran dispersos en la sustancia blanca del cerebro y la médula espinal, presumiblemente causados por el ataque de las propias células inmunitarias del cuerpo en las vainas de mielina de los procesos de las células nerviosas. Dado que los centros de desmielinización pueden ocurrir en todo el SNC, la esclerosis múltiple puede causar casi cualquier síntoma neurológico. Las alteraciones visuales y la visión borrosa pueden eventualmente provocar ceguera y trastornos de los movimientos oculares (oftalmoplejía internuclear), pero no son específicos de la esclerosis múltiple.
Junto con la epilepsia, la EM es una de las enfermedades neurológicas más comunes en los adultos jóvenes. No existe cura para la enfermedad, pero su curso a menudo puede verse influenciado favorablemente por varias medidas. La esclerosis múltiple no necesariamente conduce a discapacidades graves.
¿La EM es genética?
Aunque se desconoce la causa exacta de la EM, la mayoría de los expertos creen que una combinación de factores genéticos y ambientales actúa como desencadenante del daño característico mediado por el sistema inmunitario.
Hasta octubre de 2013, se han identificado 110 variaciones genéticas que ocurren con más frecuencia en pacientes con EM que en la población general y pueden contribuir a una predisposición a la esclerosis múltiple. Ese número ronda los 200 en 2020. Aunque cada una de estas variaciones por sí sola representa un riesgo muy bajo de desarrollo de EM, juntas representan alrededor del 20 por ciento de los componentes genéticos de la enfermedad.
Entre otras cosas, se cree que están implicados los polimorfismos de genes implicados en la vía de señalización de la interleucina. Muchas de las variantes genéticas encontradas están directamente relacionadas con el sistema inmunológico. Algunos de ellos también podrían identificarse como factores de riesgo genéticos en enfermedades autoinmunes como la diabetes tipo 1 o la enfermedad de Crohn.
De acuerdo a la Sociedad Nacional de Esclerosis Múltiple , el riesgo de la población general de desarrollar EM es de aproximadamente 1 en 750-1000. En gemelos idénticos, si uno de los gemelos tiene EM, el riesgo de que el otro gemelo desarrolle EM es de aproximadamente 1 de cada 4. El riesgo de desarrollar EM también aumenta cuando otros familiares de primer grado (padres, hermanos e hijos) tienen EM, pero mucho menos que en gemelos idénticos.
Se están realizando investigaciones para comprender mejor el riesgo genético y otros factores que contribuyen al desarrollo de la EM.
Factores de riesgo no genéticos (Parte 3 de ¿Es genética la EM?)
Se sospecha que múltiples factores no genéticos provocan la EM. Algunos de los factores más investigados se describen a continuación.
Infección
A numerosos virus (incluidos el virus de Epstein-Barr y el virus del herpes humano 6) se les ha atribuido un posible papel en el desarrollo de la enfermedad. De hecho, una reacción inmune contra el virus de Epstein-Barr es más frecuente detectable en niños con esclerosis múltiple en particular que en niños que no la padecen. Los patógenos bacterianos (que incluyen clamidia, espiroquetas, rickettsia y Streptococcus mutans) también se han asociado con el desarrollo de esclerosis múltiple.
Vitamina D
La creciente evidencia sugiere que la vitamina D juega un papel importante en la EM. Los niveles bajos de vitamina D en la sangre se han identificado como un factor de riesgo para el desarrollo de EM. Algunos investigadores creen que la exposición al sol (la fuente natural de vitamina D) puede ayudar a explicar por qué las personas que viven más cerca del ecuador tienen menos probabilidades de desarrollar EM. La teoría es que vivir más cerca del ecuador resulta en más luz solar (y por lo tanto, exposición a vitamina D) durante todo el año. La vitamina D es importante para la salud inmunológica en general.
De fumar
El papel del tabaquismo en el aumento del riesgo de EM se ha investigado durante años. Fumar antes de la aparición de la enfermedad parece aumentar significativamente el riesgo. UN metaanálisis mostró un aumento de 1,2 a 1,5 veces en el riesgo de enfermedad.
Aún no se sabe qué cambios patológicos provocados por el tabaquismo pueden influir en el desarrollo y progresión de la EM.
Obesidad
El exceso de peso en la infancia parece ser un factor en la desarrollo de la esclerosis múltiple en la edad adulta.
Microbioma
Diversos estudios han demostrado que el microbioma influye en el sistema inmunológico y en las enfermedades autoinmunes en general. Las bacterias intestinales pueden debilitar o activar el sistema inmunológico.
Epidemiología (Parte 4 de ¿Es genética la EM?)
De acuerdo a una estudiar financiado por la Sociedad Nacional de EM en 2019, casi 1 millón de personas viven con EM en los Estados Unidos. La mayoría de las personas con EM se diagnostican entre los 20 y los 50 años, aunque la EM puede ocurrir en niños pequeños y adultos mayores. Si bien los casos de EM se encuentran en la mayoría de las etnias, es más común en los caucásicos de ascendencia del norte de Europa.
Las mujeres tienen de 2 a 3 veces más probabilidades de desarrollar EM que los hombres. Se sabe que la EM ocurre con más frecuencia en áreas que están más lejos del ecuador.
Características y síntomas (Parte 5 de ¿Es genética la EM?)
Los síntomas de la EM son muy variados entre los individuos, tanto en tipo como en cantidad. Algunos de los síntomas primarios más y menos comunes de la EM son:
Síntomas comunes:
- Fatiga
- Dificultades para caminar (andar)
- Entumecimiento u hormigueo
- Espasticidad
- Debilidad
- Problemas de visión y pérdida de visión.
- Mareos y vértigo
- Problemas de vejiga
- Problemas sexuales
- Problemas intestinales
- Dolor y picazón
- Cambios cognitivos
- Cambios emocionales
- Depresión
Síntomas menos comunes:
- Problemas del habla
- Problemas para tragar
- Temblor
- Convulsiones
- Problemas respiratorios
- Pérdida de la audición

La EM tiene diferentes formas de progresión y las recaídas son frecuentes. Una recaída se define como la aparición o el resurgimiento de síntomas clínicos ya conocidos que duran más de 24 horas. Para distinguir un nuevo ataque de uno anterior, debe haber al menos 30 días entre los dos eventos clínicos.
La duración de una recaída suele ser de unos días a unas pocas semanas. Dependiendo de si los síntomas que han ocurrido retroceden por completo o solo de manera incompleta, se habla de una remisión completa o incompleta. Las llamadas pseudo recaídas ocurren en el contexto de un aumento de temperatura o junto con infecciones y pueden conducir a un empeoramiento temporal de los síntomas clínicos.
Los primeros síntomas suelen aparecer entre los 15 y los 40 años. Algunos pacientes también son un poco más jóvenes. Si bien las recaídas generalmente desaparecen por completo al comienzo de la enfermedad, los déficits neurológicos se dejan cada vez más en el curso posterior de la enfermedad después de las recaídas.
Al comienzo de la enfermedad, se observan con frecuencia alteraciones visuales y sensoriales. No es raro que la enfermedad comience con un síntoma aislado.
El síntoma que se desarrolla en una recaída depende de la localización respectiva del foco desmielinizante activo en el sistema nervioso central: por ejemplo, la inflamación en el área de las fibras del nervio óptico causa alteraciones visuales que pueden manifestarse como visión borrosa o neblina lechosa y también pueden ir acompañado de dolor en los ojos.
Un medio de cuantificar el deterioro del paciente es el uso de la Escala ampliada del estado de discapacidad (EDSS). Esta escala se utiliza para clasificar la gravedad actual de las discapacidades del paciente, con evaluación previa de las deficiencias en siete sistemas neurológicos. Al observar el curso completo de la enfermedad, son la fatiga, la disfunción de la vejiga y los trastornos del sistema motor como la parálisis y la espasticidad los que a menudo tienen el mayor impacto en la vida de los afectados.
Diagnóstico (Parte 6 de ¿Es genética la EM?)
Su médico realizará varios tipos de pruebas para diagnosticar la EM y descartar cualquier otra afección, incluidos antecedentes médicos, un examen neurológico, imágenes por resonancia magnética (IRM), análisis del líquido cefalorraquídeo y análisis de sangre para descartar otras afecciones.
A veces, los pacientes que presentan síntomas de EM serán diagnosticados con síndrome clínicamente aislado (CIS). El CIS presenta los mismos síntomas que la EM. A diferencia de la EM, un episodio de CIS es un evento singular, mientras que este último es una enfermedad de por vida. Los pacientes con cualquiera de las afecciones recibirán el mismo tratamiento.
Imágenes
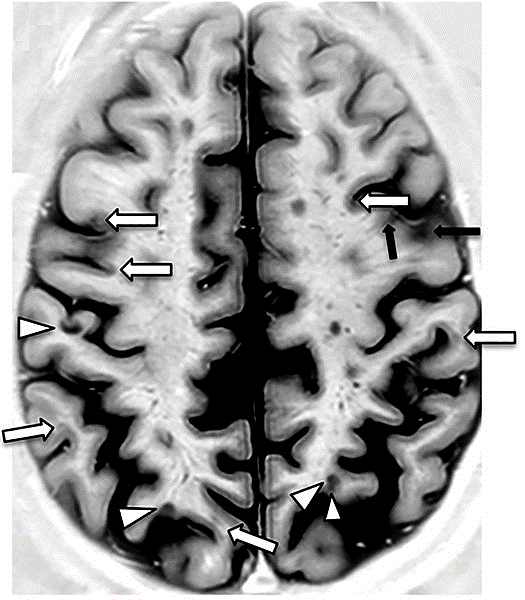
Las imágenes del cerebro y la médula espinal obtenidas por imágenes de resonancia magnética (IRM) se pueden utilizar para visualizar áreas de tejido inflamadas y cicatrizadas. Con la ayuda de un medio de contraste que contenga gadolinio, como gadopentetato de dimeglumina o ácido gadotérico, se pueden detectar focos de enfermedad aguda. Los sitios de inflamación periventriculares (alrededor de los ventrículos laterales) en el lecho medular del cerebro y los llamados sitios de haz son típicos de la EM.
Con el examen de resonancia magnética, se puede detectar la diseminación espacial y temporal de los focos inflamatorios en el cerebro y la médula espinal. Los criterios de McDonald especifican exactamente cuántos sitios de inflamación en qué región del SNC deben ser detectables para poder hablar de un resultado positivo de resonancia magnética con respecto a la diseminación espacial. El diagnóstico de EM no suele basarse únicamente en los hallazgos de las imágenes.
Otras pruebas
Punción lumbar (punción lumbar)
Se recomienda una punción lumbar en casos de sospecha de enfermedad. En el 50% de los pacientes hay un ligero aumento de células linfocíticas en el líquido cefalorraquídeo (LCR) y se detecta un proceso inflamatorio crónico en el sistema nervioso central en más del 95% de los pacientes.
Pruebas de potenciales evocados
Estas pruebas registran las señales eléctricas producidas por su sistema nervioso en respuesta a estímulos, ya sean visuales o eléctricos. En estas pruebas, usted observa un patrón visual en movimiento o se aplican impulsos eléctricos cortos a los nervios de las piernas o los brazos. Los electrodos miden qué tan rápido viaja la información a través de sus nervios.
En la mayoría de las personas con EM remitente-recidivante, se puede hacer un diagnóstico basado en un patrón de síntomas compatibles con la enfermedad y confirmado por imágenes cerebrales, como la resonancia magnética.
Para otras personas con un patrón de síntomas inusual, puede ser difícil distinguir la EM de otras enfermedades. Además de las enfermedades infecciosas, también deben excluirse otras enfermedades inflamatorias crónicas. También se deben considerar otras enfermedades inflamatorias desmielinizantes (como la neuromielitis óptica, la paraparesia espástica tropical o la encefalomielitis aguda diseminada (ADEM)). Las enfermedades del metabolismo o una deficiencia de vitamina B12 también pueden provocar síntomas similares a los de la EM.
Tratamiento (Parte 7 de ¿Es genética la EM?)
De acuerdo a la Clínica Mayo , no existe cura para la EM. El objetivo de todas las medidas terapéuticas es mantener la independencia del paciente en la vida diaria aumentando los tiempos de recuperación de los ataques, retardando la progresión de la enfermedad y controlando los síntomas de la EM.
Para tratar los ataques, los pacientes pueden usar corticosteroides recetados para reducir la inflamación del nervio o pueden optar por someterse a un intercambio de plasma, mediante el cual la sangre se extrae del cuerpo, se modifica y se reinyecta.
Para la EM primaria progresiva, el ocrelizumab (Ocrevus) es el único medicamento aprobado por la Administración de Drogas y Alimentos de los Estados Unidos (FDA) como terapia modificadora de la enfermedad. Aquellos que reciben este tratamiento tienen menos probabilidades de progresar que aquellos que no reciben tratamiento.
Hay más opciones disponibles para personas con EM remitente-recurrente. Dado que gran parte de la respuesta inmunitaria se produce al comienzo de la enfermedad, el tratamiento temprano y agresivo con estas terapias se asocia con mejores resultados.
Los tratamientos inyectables que pueden alterar los ataques del sistema inmunológico incluyen medicamentos con interferón beta y acetato de glatiramer (Copaxone, Glatopa).
Hay muchos tratamientos orales y de infusión disponibles:
Oral:
- Fingolimod (Gilenya)
- Fumarato de dimetilo (Tecfidera)
- Fumarato de diroximel (Vumerity)
- Teriflunomida (Aubagio)
- Siponimod (Mayzent)
- Cladribina (Mavenclad)
Infusión:
- Ocrelizumab (Ocrevus)
- Natalizumab (Tysabri)
- Alemtuzumab (Campath, Lemtrada)
Además de la medicación, se pueden recomendar otras estrategias para ayudar a aliviar los síntomas, especialmente en lo que respecta a los problemas musculares y de coordinación.
- Terapia física
- Relajantes musculares
- Medicamentos para reducir la fatiga.
- Medicamentos para aumentar la velocidad al caminar
- Otros medicamentos para afecciones como depresión, dolor y control de la vejiga que a menudo se asocian con la EM
¿Te gustó este artículo? Puedes encontrar más publicaciones como esta en el Biblioteca de investigación de nebulosa !