
تقرير الحمض النووي لجينوم السديم لمرض باركنسون
هل مرض باركنسون وراثي؟ أنشأنا تقرير الحمض النووي استنادًا إلى دراسة حاولت الإجابة على هذا السؤال. أدناه يمكنك رؤية تقرير SAMPLE DNA. للحصول على تقرير الحمض النووي الشخصي الخاص بك ، قم بشراء تسلسل الجينوم الكامل !

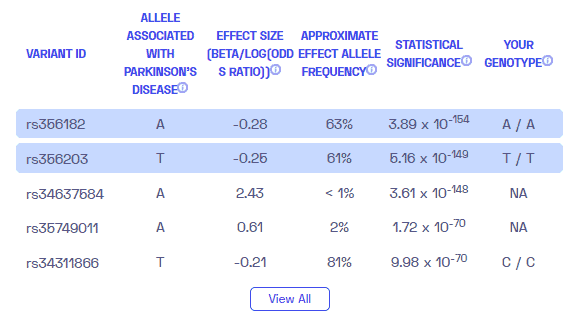
معلومة اضافية
ما هو مرض باركنسون؟ (الجزء الأول من هل مرض باركنسون وراثي؟)
ينتج مرض باركنسون أو PD عن الفقد البطيء والتدريجي للخلايا العصبية. الأعراض المميزة هي عدم القدرة على الحركة (انخفاض أو فقدان الحركة الإرادية) والصرامة والرعشة. حاليًا ، لا يوجد علاج لهذا المرض التنكسي الذي يصيب الجهاز العصبي ، على الرغم من أن الأعراض قابلة للعلاج.
يتميز مرض باركنسون بالفقدان السائد للخلايا العصبية المنتجة للدوبامين (خلايا المخ) في منطقة المادة السوداء في الدماغ. يؤدي نقص الناقل العصبي الدوبامين في النهاية إلى تقليل التأثير التنشيط للعقد القاعدية على القشرة الدماغية وبالتالي إلى اضطرابات الحركة الملحوظة.

{kind=link}
هناك عدد من التصنيفات لمرض باركنسون بناءً على أصل المرض وتطوره. تشمل هذه الفئات:
- متلازمة باركنسون مجهول السبب (IPS) (الأكثر شيوعًا)
- متلازمة باركنسون العائلية (أشكال وراثية وراثية ونادرة ، سميت باسم موضع الجين المعني (أي PARK1)
- متلازمات باركنسون العرضية (الثانوية)
- محرض بالأدوية (أي مضادات الذهان مع عداء الدوبامين)
- الشلل الرعاش الوعائي ، كما هو الحال في اعتلال الأوعية الدقيقة الدماغي (مرض بينسوانجر)
- ما بعد الصدمة (مثل اعتلال دماغ الملاكم)
- المستحثة بالسموم (مثل أول أكسيد الكربون ، المنغنيز ، MPTP)
- التهابات (على سبيل المثال ، بعد التهاب الدماغ الخمول ، وكذلك في أمراض الدماغ ذات الصلة بمسببات الأمراض المنتشرة مثل اعتلال الدماغ المتقدم بفيروس نقص المناعة البشرية)
- التمثيل الغذائي (مرض ويلسون)
- متلازمات باركنسون في سياق الأمراض العصبية التنكسية الأخرى (متلازمات باركنسون اللانمطية)
- ضمور متعدد الأجهزة
- شلل النظرات فوق النووية التقدمي
- التنكس القشري القاعدي
- خَرَف أجسام ليوي
علم الأوبئة (الجزء الثاني من هل مرض باركنسون وراثي؟)
وفقا ل مؤسسة باركنسون ، يصيب المرض أكثر من 10 ملايين شخص حول العالم. وفقًا لتوقعاتهم ، سيعيش ما يقرب من مليون مصاب بمرض باركنسون في الولايات المتحدة بحلول عام 2020 ، وهو أكثر من العدد الإجمالي للأشخاص الذين تم تشخيص إصابتهم تصلب متعدد والحثل العضلي و مرض لو جيريج (أو التصلب الجانبي الضموري).
يتم تشخيص ما يقرب من 60 ألف أمريكي بمرض باركنسون كل عام.

{kind=link}
تزداد نسبة الإصابة بمرض باركنسون مع تقدم العمر ، ولكن يُشخص ما يقدر بنحو أربعة بالمائة من المصابين بهذا المرض قبل سن الخمسين. الرجال أكثر عرضة للإصابة بمرض باركنسون 1.5 مرة أكثر من النساء.
الأعراض (الجزء 3 من هل مرض باركنسون وراثي؟)
يبدأ المرض تدريجياً ويتطور طوال الحياة. تصبح الأعراض أكثر حدة مع تقدم المرض وبالتالي يسهل التعرف عليها. عادةً ما يبدأ الشكل الأكثر شيوعًا لمرض باركنسون من جانب واحد ، في جانب واحد من الجسم. لذلك ليس من غير المألوف حدوث آلام في الكتف وتوتر عضلي من جانب واحد ، مما يدفع المريض إلى زيارة طبيب العظام أولاً.
وفقا ل مؤسسة باركنسون عشرة أعراض مبكرة تشمل:
- رعشه
- خط صغير
- فقدان حاسة الشم
- مشاكل في النوم
- صعوبة في الحركة أو المشي
- إمساك
- صوت رقيق أو منخفض
- وجه مقنع
- دوار أو إغماء
- الانحناء أو الانحناء
مع تقدم المرض ، يتم تعريف مرض باركنسون من خلال الأعراض الأساسية لبطء الحركة (الحركة البطيئة) أو عدم القدرة على الحركة (فقدان أو ضعف قوة الحركة الإرادية) وأحد الأعراض الثلاثة الرئيسية الأخرى (الصلابة ، والرعشة ، وعدم استقرار الوضع).
أكينيسيا و / أو بطء الحركة
هذا النقص العام في الحركة هو شرط أساسي لتشخيص مرض باركنسون. يمكن ملاحظته في جميع الحركات. وبالتالي ، تقل حركة العضلات في الوجه ، ويصبح الكلام هادئًا وغير واضح ، ويتأخر البلع ، وتقل براعة اليدين خاصة مع الحركات السريعة (يصبح خط اليد أصغر) ، وتصبح حركات الجذع صعبة ، وتصبح المشية صغيرة. والخلط.
في الشلل الرعاش ، يحدث هذا البطء بطرق مختلفة:
- تقليل الحركات التلقائية (مثل الوميض أو تأرجح ذراعيك عند المشي)
- صعوبة في بدء الحركات (مثل النهوض من الكرسي)
- البطء العام في الأفعال الجسدية
- ظهور سكون غير طبيعي أو انخفاض في تعبيرات الوجه
يترجم هذا إلى صعوبة أداء الوظائف اليومية ، مثل زر قميص أو تقطيع الطعام أو تنظيف أسنانك.
الاستعلاء
يشير هذا إلى تصلب العضلات بسبب زيادة توتر العضلات. وهو ناتج عن توتر لا إرادي للعضلات المخططة بالكامل وغالبًا ما يؤدي أيضًا إلى آلام في العضلات. يظهر ظاهريًا ثنيًا طفيفًا في مفصل الكوع والجذع والرقبة ، وبعد ذلك في مفاصل الركبة.
رعشه
ينتج عن التوتر المتناوب للعضلات المتعارضة رعشة بطيئة نسبيًا تقل مع الحركة. يكون الرعاش أكثر بروزًا في جانب واحد من الجسم.
عدم الاستقرار الوضعي
يحدث انخفاض الاستقرار في تثبيت الجسم في وضع مستقيم بسبب اضطراب في ردود الفعل الموضعية. تتأخر الحركات التعويضية الصغيرة ولكن السريعة ، مما يؤدي إلى عدم استقرار المشية والوقوف. تصبح حركة الدوران غير مستقرة وقد يبدأ المرضى في التعثر.
يمكن أن تظهر الأعراض المختلفة بدرجات مختلفة في المرضى الأفراد أو تكون غائبة تمامًا. كما يتغير حدوثها وشدتها خلال اليوم.

{kind=link}
قد يعاني المرضى أيضًا من أمراض أخرى مثل انخفاض ضغط الدم ومشاكل النوم.
الأسباب (الجزء 4 من هل مرض باركنسون وراثي؟)
يعتقد العلماء أن مجموعة من العوامل الوراثية والبيئية تلعب دورًا في فقدان الدوبامين في الدماغ والظهور التالي لمرض باركنسون. المرض متنوع للغاية بين المرضى ، مع اختلاف الأفراد في العمر ، والتقدم ، وحتى فعالية العلاج.
نقص الدوبامين
مرض باركنسون هو مرض تنكسي يصيب الجهاز الحركي خارج الهرمي (EPS) أو العقد القاعدية. إنه ينطوي على موت الخلايا العصبية في الجزء المضغوط من المادة السوداء ، والتي تنتج الدوبامين وتنقله عبر محاورها إلى البوتامين. يتم ملاحظة العلامات الأولى للمرض فقط عند التقريب. مات 55٪ إلى 60٪ من خلايا الدوبامين هذه.
يؤدي نقص الدوبامين في النهاية إلى خلل في وظيفة العقد القاعدية بطريقتين. مادة الجلوتامات الرسول وفيرة نسبيًا. يثبط globus pallidus internus في النهاية التنشيط الحركي للقشرة الدماغية بواسطة المهاد. يؤدي هذا إلى الأعراض الرئيسية الشديدة والرعشة ونقص الحركة ، ولكن أيضًا إلى تباطؤ العمليات العقلية (بطء الشخصية).
بالإضافة إلى نقص الدوبامين ، لوحظت أيضًا تغيرات في النواقل العصبية الأخرى. على سبيل المثال ، تم العثور على نقص السيروتونين والأسيتيل كولين والنورادرينالين في بعض مناطق جذع الدماغ.
هل مرض باركنسون وراثي؟
أظهر البحث والتعرف على الأشكال الموروثة أن مرض باركنسون ليس مرضًا موحدًا ، ولكنه مجموعة غير متجانسة من الأمراض مع مجموعة من المظاهر السريرية والمرضية (PARK1 إلى PARK13). تاريخ العائلة هو عامل خطر مهم. الأشكال أحادية الجين من مرض باركنسون مسؤولة عن حوالي خمسة إلى عشرة بالمائة من جميع المرضى الذين يصابون بمرض باركنسون. من بين هذه الطفرات الجينية لجين ألفا سينوكلين (جين SNCA ، PARK1) ذات أهمية خاصة.
تم تحديد موقع PARK1 في عائلة كبيرة مصابة بمرض باركنسون الوراثي السائد وأمراض جسم ليوي ؛ تم تحديد اثنين من الطفرات النقطية الأخرى ذات الاختراق العالي في العائلات الكبيرة ولكن ليس في المرضى الذين يعانون من مرض باركنسون المتقطع.
بشكل ملحوظ ، في عام 2007 ، تم تحديد مجاميع SNCA في جزء معين من الدماغ يسمى الجزء قبل المشبكي من أنسجة المخ البشري. ربما كتعبير عن الخلل الوظيفي التشابكي المبكر ، على الرغم من أن العلاقة الدقيقة بين التجميع والخلل الخلوي وموت الخلايا لم تعرف بعد.
بالإضافة إلى التغييرات في تسلسل الأحماض الأمينية ، فإن الازدواجية والمضاعفات تؤدي أيضًا إلى زيادة ميل البروتين لتشكيل أوليغومرات ومجموعات ليفية ، بحيث يلعب تنظيم تعبير وترجمة SNCA دورًا مهمًا على الأقل.
يُعتقد أن الجينات تسبب حوالي 10 إلى 15 في المائة من جميع حالات مرض باركنسون والأشخاص الذين يعانون من أفراد الأسرة المصابين بمرض باركنسون معرضون بشكل أكبر للإصابة بالمرض عادة ما يرتبط البداية المبكرة بنسخ متعددة من الجينات المرتبطة بالمرض. تشمل الطفرات الأكثر شيوعًا في الجينات المرتبطة بمرض باركنسون LRRK2 و GBA و SNCA.
LRRK2: حدثت طفرات في جين LRRK2 مبين لتغيير الخلايا العصبية. يمكن العثور على التغيرات الجينية في هذا الجين في ما يصل إلى 2٪ من جميع المصابين بمرض باركنسون. هناك على الأقل 20 طفرة مختلفة التي يمكن أن تحدث في هذا الجين ، بعضها أكثر شيوعًا ، مثل G2019S الذي يحدث بشكل أكثر تكرارا في عائلات شمال إفريقيا.
GBA: يقوم هذا الجين بترميز بروتين يشارك في إزالة النفايات الخلوية من الخلايا. ما بين 5 إلى 10٪ من المصابين بمرض باركنسون لديهم طفرة في هذا الجين. ومع ذلك ، فإن فرص إصابة أولئك الذين يحملون هذه الطفرة بمرض باركنسون في المستقبل منخفضة نسبيًا. تم ربط عدد قليل فقط من الطفرات في جين GBA بزيادة خطر الإصابة بمرض باركنسون.
SNCA: يرتبط هذا الجين بمرض باركنسون في أنه ينتج بروتين ألفا سينوكلين. تحتوي أدمغة المصابين بمرض باركنسون على كتل من هذا البروتين تسمى أجسام ليوي. يُعتقد أن الطفرات في جين SNCA يمكن أن تسبب كمية زائدة من هذا البروتين في أدمغتهم ، والتي تشكل بعد ذلك أجسام ليوي وتصبح سامة.
دراسة مؤسسة باركنسون ، جينات PD : رسم خريطة لمستقبل مرض باركنسون ، هو أول دراسة وطنية تقدم الاختبارات الجينية المنزلية والاستشارات دون تكلفة لأولئك الذين لديهم تشخيص مؤكد لمرض باركنسون. يمكن للمرضى أيضًا إرسال النتائج من مواقع الاختبارات الجينية الأخرى مثل علم جينوم السديم ، والذي يقدم تسلسل الجينوم الكامل 30X.
عادةً ما يبحث الاختبار الجيني لمرض باركنسون عن الطفرات في الجينات: GBA و PARK7 و SNCA و LRRK2 و parkin و PINK1.
العوامل البيئية
بالإضافة إلى العوامل الوراثية ، قد تؤدي العديد من عوامل الخطر البيئية إلى زيادة خطر الإصابة بمرض باركنسون ، على الرغم من أن الدراسات العلمية لتأكيد هذه الارتباطات كانت غير متسقة إلى حد كبير. تشمل هذه العوامل:
- إصابة بالرأس
- التعرض للمعادن
- التعرض للمذيبات وثنائي الفينيل متعدد الكلور (PCBs)
من ناحية أخرى ، فقد ظهر ارتباط قوي بين شلل الرعاش والتعرض لمبيدات الآفات ومبيدات الأعشاب. أحد هذه المبيدات هو الباراكوات ، وهو مبيد أعشاب تجاري يستخدم في الولايات المتحدة محظور في 32 دولة ، بما في ذلك الاتحاد الأوروبي والصين.
التشخيص (الجزء الخامس من هل مرض باركنسون وراثي؟)
لا يوجد اختبار محدد لتشخيص مرض باركنسون ، وبالتالي فإن التشخيص يعتمد فقط على التاريخ الطبي والأعراض والفحص العصبي. قد يكون من الصعب تشخيص مرض باركنسون في مراحله المبكرة ، عندما تكون الأعراض ضئيلة. في حين يمكن إجراء التشخيص الأولي من قبل أي مقدم طبي ، يتم التأكيد عادة من قبل طبيب أعصاب مدرب. يجب أن يكون هناك اثنان من الأعراض الأربعة الرئيسية خلال فترة زمنية:
- اهتزاز أو رعاش
- بطء في الحركة يسمى بطء الحركة
- تصلب أو تصلب الذراعين أو الساقين أو الجذع
- مشاكل في التوازن واحتمالية السقوط ، وتسمى أيضًا عدم استقرار الوضع
قد يتم إجراء معظم الاختبارات مثل اختبارات الدم واختبارات التصوير لاستبعاد الحالات الأخرى التي قد تسبب الأعراض.
العلاج (الجزء 6 من هل مرض باركنسون وراثي؟)
بالنسبة الى مايو كلينيك ، يعتمد العلاج على الأعراض الفردية. تتضمن طرق علاج مرض باركنسون مجموعة واحدة أو مجموعة من الأدوية والجراحة وتعديلات نمط الحياة ، مثل العلاج الطبيعي والتمارين الرياضية. من المهم ملاحظة أن الأدوية تُستخدم للتحكم في الأعراض وليس علاج المرض.
الأدوية
تعمل معظم الأدوية على زيادة أو استبدال أو حماية الدوبامين في الدماغ.
كاربيدوبا ليفودوبا: الدواء الأكثر شيوعًا هو L-dopa (levodopa) ، وهو مقدمة للدوبامين. هذا السلائف ، على عكس الدوبامين نفسه ، قادر على عبور الحاجز الدموي الدماغي ويتحول إلى دوبامين في الدماغ. يتم دمج Levodopa مع carbidopa (Lodosyn) ، والذي يحمي ليفودوبا من التحول المبكر إلى الدوبامين
تشمل الأشكال الأحدث من ليفودوبا Inbrija (الشكل المستنشق) و Duopa (شكل التسريب).
ناهضات الدوبامين: بعد عدة سنوات من تناول L-dopa ، يمكن أن تحدث حركات لا إرادية. لهذا السبب ، يُنصح عادةً بالعلاج بمنشطات الدوبامين طويلة المفعول في بداية مرض باركنسون ، خاصةً في المرضى الأصغر سنًا. بدلاً من التحول إلى الدوبامين ، تعتبر ناهضات الدوبامين عقاقير مختلفة تحاكي عمل الدوبامين وتحفز مستقبلات الدوبامين.
على الرغم من أنها ليست فعالة مثل ليفودوبا ، إلا أنها تدوم لفترة أطول
تشمل ناهضات الدوبامين براميبيكسول (ميرابيكس) ، روبينيرول (ريكويب) وروتيجوتين (نيوبرو ، يُعطى على شكل رقعة). Apomorphine (Apokyn) هو ناهض للدوبامين قابل للحقن قصير المفعول يستخدم للتخفيف السريع.
مثبطات MAO B: يساعد هذا الدواء في منع انهيار الدوبامين عن طريق تثبيط إنزيم يعمل على استقلاب الدوبامين في الدماغ.
مثبطات COMT: يطيل هذا الدواء بشكل معتدل من تأثير ليفودوبا عن طريق منع الإنزيم الذي يكسر الدوبامين. عندما تؤخذ مع ليفودوبا ، فإنها تزيد من توافر الليفودوبا بنسبة 40 إلى 90 في المائة وتطيل عمر النصف في البلازما. تشمل الأدوية في هذه الفئة entacapone و tolcapone.
أدوية أخرى: الأدوية الأخرى التي يتم استخدامها بشكل نادر هي مضادات الكولين والأمانتادين. كانت مضادات الكولين توصف في السابق للمساعدة في السيطرة على الرعاش. ومع ذلك ، فإن الآثار الجانبية ، بما في ذلك ضعف الذاكرة ، والارتباك ، والهلوسة ، والإمساك ، وجفاف الفم وضعف التبول ، أدت إلى عدم وصفها في كثير من الأحيان. قد يوفر أمانتادين راحة قصيرة المدى ولكنه يؤدي أيضًا إلى بعض الآثار الجانبية الخطيرة مثل التبقع الأرجواني في الجلد وتورم الكاحل والهلوسة.

{kind=link}
جراحة
التحفيز العميق للدماغ هو إجراء جراحي قد يساعد في تخفيف بعض أعراض مرض باركنسون. يقوم الجراحون بزرع أقطاب كهربائية في جزء معين من دماغك. يتم توصيل الأقطاب الكهربائية بمولد مزروع في صدرك. تُستخدم النبضات الكهربائية المرسلة إلى عقلك لتقليل أعراض المرض.
مثل أي عملية جراحية ، ينطوي التحفيز العميق للدماغ على مخاطر ، بما في ذلك العدوى والسكتات الدماغية ونزيف الدماغ. وقد يكون من الضروري إجراء تعديلات على النظام إذا لم يستجيب المريض جيدًا للتنبيه.
غالبًا ما يتم تقديم التحفيز العميق للدماغ للأشخاص المصابين بمرض باركنسون المتقدم والذين لديهم استجابات دوائية غير مستقرة. هو الأكثر فائدة للسيطرة على الرعاش. يمكن أن يؤدي التحفيز العميق للدماغ إلى استقرار تقلبات الأدوية وتقليل أو إيقاف الحركات اللاإرادية (خلل الحركة) وتقليل الرعاش وتقليل الصلابة وتحسين تباطؤ الحركة.
للحصول على معلومات إضافية ومعلومات التجارب السريرية وخيارات العلاج ، قم بزيارة مؤسسة باركنسون.
إذا أعجبك هذا المقال ، فيجب عليك التحقق من منشوراتنا الأخرى في مكتبة أبحاث السديم !