Informe de ADN de Nebula Genomics para ELA
¿La ELA es genética? Creamos un informe de ADN basado en un estudio que intentó responder esta pregunta. A continuación puede ver un informe de MUESTRA de ADN. Para obtener su informe de ADN personalizado, compre nuestro Secuenciación del genoma completo !

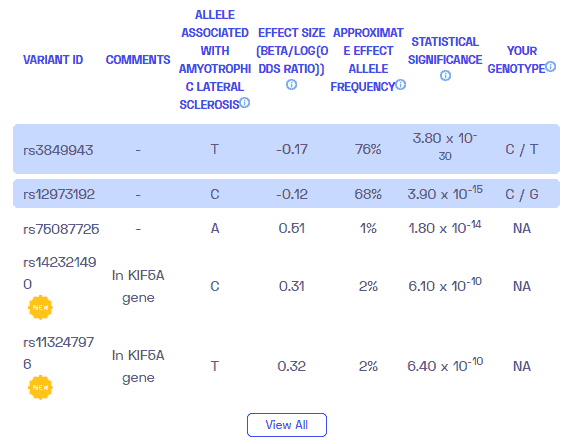
Información Adicional
¿Qué es la ELA? (Parte 1 de ¿La ELA es genética?)
Esclerosis lateral amiotrófica (ELA) Pertenece al grupo de las enfermedades de las neuronas motoras y es una enfermedad degenerativa rara del sistema nervioso motor. Afecta negativamente a los nervios y las neuronas motoras. Por tanto, cualquier actividad que requiera el uso de músculos voluntarios, como caminar, hablar o masticar, se debilita gradualmente con el tiempo en esta enfermedad. Esto conduce a una limitación progresiva en las actividades de la vida diaria. Aunque no existe cura para la ELA, la rapidez con la que progresa la enfermedad puede ralentizarse.
Otros nombres de la enfermedad son síndrome de Lou Gehrig o enfermedad de Charcot. Este último fue el descriptor de la condición, Jean-Martin Charcot. Lou Gehrig era un jugador de béisbol de los Yankees de Nueva York y tuvo que retirarse después de ser diagnosticado con ELA. El destino de Gehrig hizo que la rara enfermedad fuera conocida por un gran público por primera vez. El famoso Ice Bucket Challenge de 2014 fue fundamental para recaudar fondos para la investigación de ALS (Fuente de información: Asociación ALS: Desafío del cubo de hielo ).
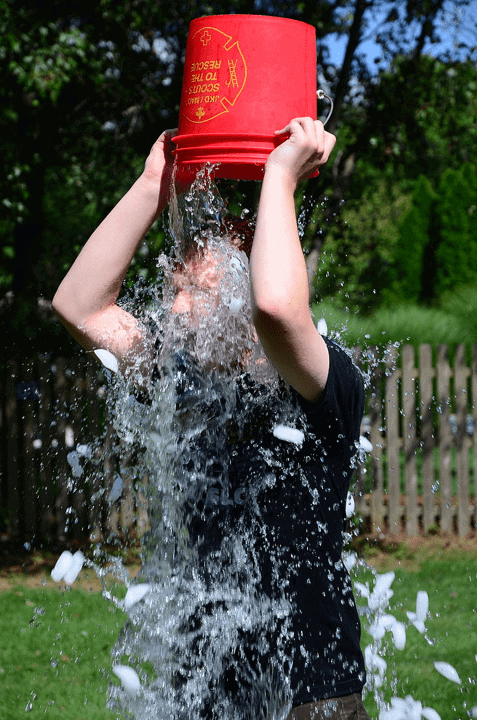
.jpg){kind=link}
Se están llevando a cabo investigaciones sobre las causas y los posibles tratamientos de la enfermedad. Organizaciones nacionales como Instituto de Desarrollo de Terapia ALS (TDI) , la Asociación ALS , y el Centros para el Control y la Prevención de Enfermedades (CDC) todos han financiado proyectos en esta área en 2020.
Epidemiología (Parte 2 de ¿La ELA es genética?)
A partir de 2014, el Centros para el Control y la Prevención de Enfermedades ha informado de 16.000 casos de ELA en los Estados Unidos. No se conoce mucho un registro exacto de sus estadísticas muy recientes. En los EE. UU., Alrededor de 5-7 existen casos por cada población de 1,00,000. Es en el rango de edad de 55 a 75 años cuando finalmente se diagnostica la enfermedad. Sin embargo, vale la pena señalar que la afección rara vez afecta a pacientes más jóvenes entre los 25 y los 35 años. También es más común en hombres que en mujeres (la proporción de género es de aproximadamente 1,5: 1).
Formas (Parte 3 de ¿La ELA es genética?)
Principalmente hay tres formas de ELA.
ELA esporádica : Este tipo ocurre al azar sin ninguna causa probable. La mayoría (90 a 95%) de los casos de ELA son esporádicos. Se sospecha que un combinación de factores genéticos y ambientales son responsables.
Familiar : En los EE. UU., La forma familiar de la enfermedad se observa en una minoría de los casos (5 a 10%), donde las personas heredan ELA de un miembro de la familia. La mayoría de los casos se heredan con un patrón autosómico dominante, lo que significa que una copia del gen alterado en cada célula es suficiente para causar el trastorno.
Guameño : Se ha observado una forma guameña de la enfermedad en la población de Guam, donde la causa es el consumo de palma sagú falsa, Cycas micronesia.
Fisiopatología de la ELA (Parte 4 de ¿Es genética la ELA?)
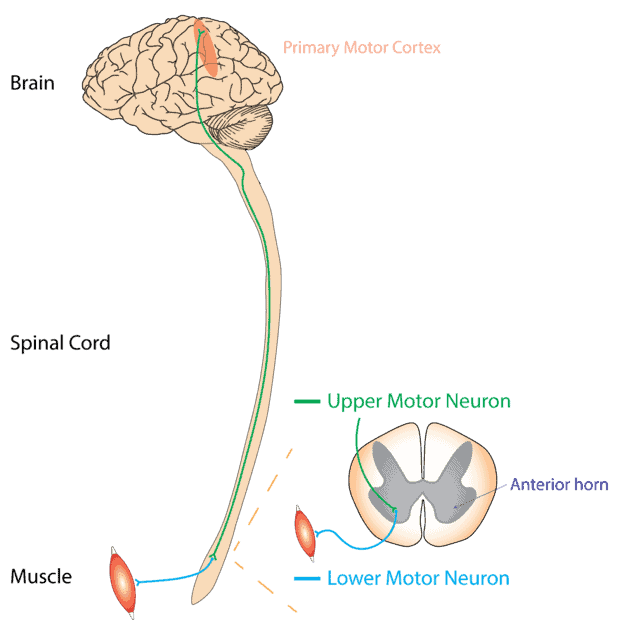
En la ELA, las neuronas motoras superiores e inferiores del cerebro y la médula espinal se ven afectadas. Las neuronas motoras superiores ocupan el cerebro mientras que las neuronas motoras inferiores están en la médula espinal. Según el sitio web de ALS Pathways, generalmente se propaga desde un punto focal, interrumpiendo así todas las neuronas motoras del cuerpo y, en última instancia, causando la muerte de las neuronas motoras. Y múltiples factores son responsables de este tipo de enfermedad neurodegenerativa. Otras formas pueden afectar solo a las neuronas motoras superiores (esclerosis lateral primaria, PLS) o las neuronas motoras inferiores (atrofia muscular progresiva, PMA).
La degeneración de las neuronas motoras superiores conduce a un aumento del tono muscular o parálisis espástica. Mientras que el daño de la neurona motora inferior conduce a un aumento de la debilidad muscular. Esto interfiere con el correcto funcionamiento de la actividad diaria.
{kind=link}
Causas (Parte 5 de ¿La ELA es genética?)
En la ELA, se sospecha que los genes influyen en el riesgo de desarrollar la enfermedad. Los estudios respaldan firmemente los factores genéticos relacionados con la ELA. Sin embargo, los estudios que implican el papel de algunos de los factores ambientales no son muy consistentes. En la mayoría de los casos, se desconoce la causa exacta. Pero en el resto de la pequeña cohorte (5-10%), los factores genéticos pueden explicar la afección. Otros factores como los antecedentes familiares, el género, la geografía, etc. también pueden tener asociaciones con la ELA.
¿La ELA es genética?
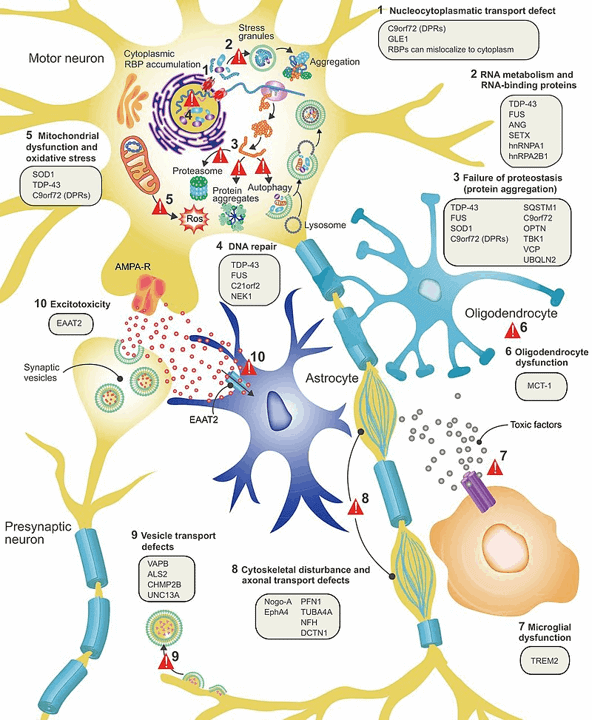
Varias variantes genéticas están estrechamente relacionadas con la ELA. Las mutaciones genéticas específicas asociadas con la enfermedad conducen a la acumulación patológica o la degradación prematura de proteínas mal plegadas que finalmente desencadenan la neurodegeneración.
Principales componentes genéticos de la ELA
El gen SOD1 codifica una enzima conocida como superóxido dismutasa. Esta enzima juega un papel importante en el mecanismo anti-defensa. Las variantes mal plegadas de las proteínas SOD1 conducen a la progresión.
Este gen codifica TDP-43, una proteína que regula la expresión génica. TDP-43 es crucial para varias etapas del procesamiento de ARN. Algunas mutaciones genéticas dominantes en TARDBP dan como resultado un TDP-43 defectuoso, lo que causa neurodegeneración. Por tanto, los cambios en la distribución y las funciones de esta proteína tienen una fuerte asociación con la ELA.
El gen FUS expresa la proteína FUS, que es un tipo de proteína de unión al ARN. Algunas de sus funciones son similares a las de la proteína TDP-43. Por lo tanto, la proteína FUS monitorea una serie de procesos en el metabolismo del ARN. En la ELA, las variantes patogénicas de este gen a veces provocan un plegamiento y una distribución inadecuados de la proteína FUS, lo que interfiere con la fisiología celular normal.
C9ORF72: aunque el papel exacto de este gen aún no está claro, pocos informes han indicado su participación en el transporte de proteínas, especialmente en forma de actividad endosómica y autofagia. Y una expresión disminuida de C9ORF72 podría estar asociada con una enfermedad, como se señaló en algunos estudios en animales. Sin embargo, esta regulación a la baja por sí sola no da como resultado ELA, pero de hecho, funciona en un complejo de algunas otras proteínas.
Componentes genéticos menores de la ELA
Pocos otros modificadores genéticos en la ELA son ATXN1, ATXN2, UNC13A. Las mutaciones en estos genes probablemente hagan que las personas susceptible al desorden. Pero no se correlacionan fuertemente con esta enfermedad.
Componente no genético de la ELA (Parte 7 de ¿Es genética la ELA?)
Factor de riesgo laboral
Un numero de informes tener propuesto que los atletas profesionales tienen más probabilidades de desarrollar ELA. Y esto podría deberse principalmente a lesiones graves en la cabeza o actividades físicas intensas.
Otros ejemplos de exposición laboral Son casos en los que las personas están expuestas a campos magnéticos o trabajos físicos extenuantes. Sin embargo, estos estudios se basan en asociaciones, no son consistentes y tienen una etiología desconocida.
Químicos tóxicos
Los resultados de un estudiar en la Universidad de Michigan en Ann Arbor (mayo de 2016) confirmaron la sospecha de larga data de que la exposición a toxinas ambientales puede aumentar el riesgo de desarrollar ELA. En este estudio, los investigadores observaron una asociación significativa entre el trastorno y la presencia de hidrocarburos clorados, bifenilos policlorados y bifenilos polibromados.
Beta metilamino L-alanina (BMAA)
BMAA es una neurotoxina asociada con ELA. Según los informes, una población indígena que vive en Guam tiene un mayor riesgo de contraer la enfermedad. La dieta de esta población es rica en BMAA. Generalmente, las cianobacterias en el agua de las islas del Pacífico producen BMAA. Y dado que la gente de Guam depende de los productos del mar, esto puede poner a la población en un mayor riesgo .
Microbioma
El microbioma intestinal a menudo influencias el resultado de la enfermedad, como se informó en estudios con animales. Microbioma intestinal estudios en ratones han demostrado que existe un vínculo importante entre algunas de las cepas bacterianas y la ELA. Además, en este estudio, una cepa de Akkermansia muciniphila tiende a ralentizar el progreso de la enfermedad en ratones. Otro Estudio 2020 en ratones ha informado que las comunidades microbianas intestinales influyen en el resultado de la supervivencia. Esto se observó especialmente, porque los ratones susceptibles a la ELA, a pesar de tener antecedentes genéticos similares, mostraron diferencias significativas en la esperanza de vida. Los patrones de diversidad de las comunidades microbianas fueron significativamente diferentes, uno de los factores que podría haber contribuido a las diferencias en la supervivencia de esos ratones.
Síntomas (Parte 8 de ¿La ELA es genética?)
Los síntomas característicos de la ELA son el deterioro funcional del movimiento muscular en las extremidades superiores e inferiores, los músculos bulbares y los músculos del tronco.
De acuerdo con la Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares , los síntomas se acumulan gradualmente. Algunos de los primeros síntomas de la ELA incluyen:
- Fasciculaciones (espasmos musculares) en el brazo, la pierna, el hombro o la lengua
- Calambres musculares
- Músculos tensos y rígidos (espasticidad)
- Debilidad muscular que afecta un brazo, una pierna, el cuello o el diafragma.
- Habla arrastrada y nasal
- Dificultad para masticar o tragar
Dependiendo de los primeros síntomas observados, la afección se puede dividir en subcategorías. Cuando los síntomas comienzan por primera vez en los brazos o las piernas, se conoce como ELA de «aparición en las extremidades». Si los primeros indicios son problemas del habla o de la deglución, se denomina ELA de “inicio bulbar”.
La ELA progresa de manera diferente en diferentes individuos. Sin embargo, en todos los casos, la debilidad muscular y la atrofia se extienden a otras partes del cuerpo hasta que el funcionamiento se vuelve difícil. Las personas con etapas avanzadas de la enfermedad eventualmente no podrán pararse, caminar o usar sus extremidades y es posible que les resulte difícil tragar, hablar y respirar.
En la mayoría de los casos, las personas con ELA conservan su capacidad para realizar procesos mentales superiores como el razonamiento, el recuerdo, la comprensión y la resolución de problemas.
El tiempo medio de supervivencia del paciente es de unos tres a cinco años desde el inicio de la enfermedad, sin embargo, 10-20 por ciento de los pacientes pueden sobrevivir durante más tiempo.
Diagnóstico (Parte 9 de ¿La ELA es genética?)
En su mayoría, los síntomas varían de una persona a otra. Por lo tanto, un médico realizará un examen completo utilizando múltiples pruebas . A partir de ahora, no existe ningún Prueba genética para ELA.
Examen físico: En la mayoría de los casos, el aumento de la debilidad muscular, la atrofia muscular o incluso los espasmos musculares llevan a los pacientes a consultar al médico por primera vez. Cabe señalar que las contracciones musculares solas casi siempre representan fasciculaciones benignas. Al principio, los síntomas pueden aparecer en cualquier parte del cuerpo. Durante la exploración, la aparición simultánea de signos de parálisis flácida y espástica refuerza la sospecha del diagnóstico de ELA.
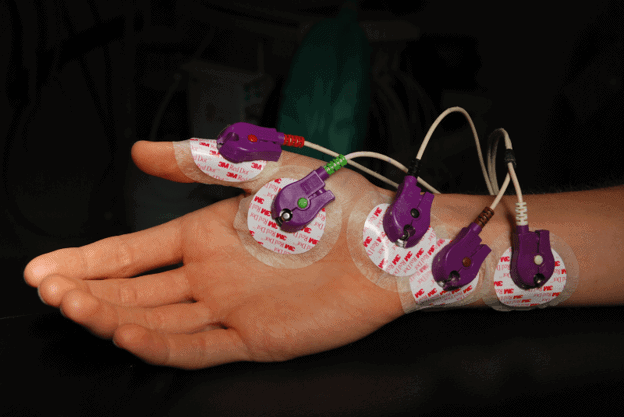
Electromiografía: Un examen electromiográfico y electroneurográfico es indispensable para hacer un diagnóstico. La electromiografía analiza la actividad eléctrica en los músculos. Para la realización del diagnóstico se han desarrollado criterios estandarizados internacionalmente (criterios de El Escorial). De acuerdo con estos criterios, un diagnóstico requiere examinar la degeneración de las neuronas motoras superiores e inferiores, su evolución y también la ausencia de cualquier otra enfermedad neurodegenerativa.
Punción lumbar: El médico extrae un pequeño volumen de líquido cefalorraquídeo y busca la presencia de afecciones inflamatorias o células anormales. Esta prueba es importante solo en casos con síntomas inusuales.
Biopsia: Generalmente, un cirujano extrae una parte del músculo de la parte superior del brazo o del muslo. Luego, el cirujano busca cualquier daño mediante tinción y microscopía.
Tratamiento (Parte 10 de ¿La ELA es genética?)
Por lo general, el tratamiento comprende una combinación de terapia con medicamentos y terapia orientada a los síntomas. La terapia sintomática tiene como objetivo ayudar a evitar las complicaciones de la debilidad muscular y mejorar la calidad de vida del paciente. En modelos animales y células madre humanas, genético estudios han identificado posibles dianas farmacológicas, lo que podría ser un paso adelante hacia el desarrollo de la farmacoterapia.
A veces, las personas pueden querer someterse a pruebas genéticas, especialmente en casos con antecedentes familiares de ELA. Luego, en tales casos, el asesor genético puede ayudar a evaluar no solo los antecedentes familiares, sino también los antecedentes médicos y cualquier otro riesgo potencial. Para la ELA familiar, generalmente se observa un resultado positivo en 60-70 por ciento de los casos.
Terapia neuroprotectora
El riluzol es el fármaco común en la terapia neuroprotectora. Este fármaco inhibe la destrucción de las células nerviosas motoras al bloquear la liberación del neurotransmisor glutamato de las neuronas.
Se informa que la administración de riluzol prolonga la supervivencia en aproximadamente tres meses y prolonga las etapas iniciales de la ELA. Sin embargo, debe tenerse en cuenta que el riluzol no puede detener completamente la progresión de la enfermedad.
A partir de 2017, la Administración de Drogas y Alimentos de los EE. UU. Ha otorgado la aprobación de otro medicamento, Edaravone. Indicativamente ralentiza el desarrollo de ELA.
Terapia de síntomas
Los siguientes síntomas a menudo se tratan con terapias adecuadas:
- Calambres musculares
- Disfagia
- Salivación
- Trastorno del habla y comunicación
- Risa y llanto patológicos
- Desórdenes respiratorios
- Depresión, trastornos del sueño y ansiedad.
Noticias recientes (2020)
Los investigadores del Trinity College Dublin han iniciado el Ensayo clínico de fase 1 de terapia génica en el Centro de Investigación Clínica, St. James Hospital Dublin (a partir del 1 de septiembre de 2020). Esta terapia basada en genes se dirige al gen C9ORF72. La compañía farmacéutica estadounidense Biogen ha patrocinado este ensayo clínico.
¿Te gustó este artículo? Asegúrese de revisar las otras publicaciones en el Biblioteca de investigación de nebulosa !