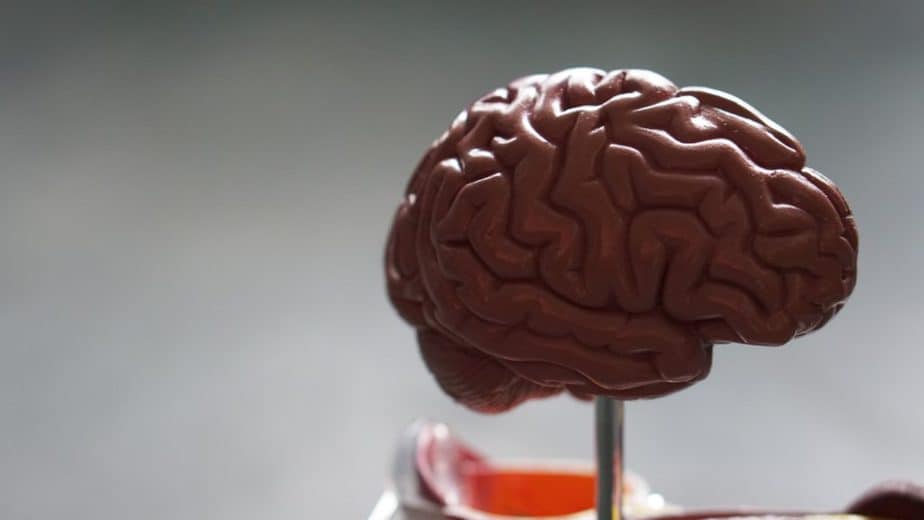
Rapport ADN Nebula Genomics pour la SLA
La SLA est-elle génétique? Nous avons créé un rapport ADN basé sur une étude qui a tenté de répondre à cette question. Ci-dessous vous pouvez voir un exemple de rapport ADN. Pour obtenir votre rapport ADN personnalisé, achetez notre Séquençage du génome entier !

Information additionnelle
Qu’est-ce que la SLA? (Partie 1 de La SLA est-elle génétique?)
Sclérose latérale amyotrophique (SLA) appartient au groupe des maladies des motoneurones et est une maladie dégénérative rare du système nerveux moteur. Il affecte négativement les nerfs et les motoneurones. Par conséquent, toute activité qui nécessite l’utilisation de muscles volontaires, comme la marche, la parole ou la mastication, s’affaiblit progressivement avec le temps dans cette maladie. Cela conduit à une limitation progressive des activités de la vie quotidienne. Bien qu’il n’existe aucun remède contre la SLA, la vitesse à laquelle la maladie progresse peut être ralentie.
Les autres noms de la maladie sont le syndrome de Lou Gehrig ou la maladie de Charcot. Ce dernier était le descripteur de la condition, Jean-Martin Charcot. Lou Gehrig était un joueur de baseball des Yankees de New York et a dû prendre sa retraite après avoir reçu un diagnostic de SLA. Le sort de Gehrig a fait connaître la maladie rare à un large public pour la première fois. Le célèbre Ice Bucket Challenge de 2014 a contribué à collecter des fonds pour la recherche sur la SLA (Source d’information: ALS Association: Défi du seau de glace ).
.jpg){kind=link}
Des recherches sur les causes et les traitements potentiels de la maladie sont en cours. Des organisations nationales telles que Institut de développement thérapeutique de la SLA (TDI) , la Association ALS , et le Centres de contrôle et de prévention des maladies (CDC) tous ont financé des projets dans ce domaine en 2020.
Épidémiologie (partie 2 de La SLA est-elle génétique?)
Depuis 2014, le CDC a signalé 16 000 cas de SLA aux États-Unis. Un enregistrement exact de ses statistiques très récentes est peu connu. Aux États-Unis, environ 5-7 des cas existent pour chaque population de 1,00,000. C’est entre 55 et 75 ans que la maladie est finalement diagnostiquée. Cependant, il convient de noter que la maladie affecte rarement les patients plus jeunes entre 25 et 35 ans. Il est également plus fréquent chez les hommes que chez les femmes (le rapport entre les sexes est d’environ 1,5: 1).
Formulaires (partie 3 de La SLA est-elle génétique?)
Il y a principalement trois formes de la SLA.
SLA sporadique : Ce type se produit au hasard sans aucune cause probable. Une majorité (90 à 95%) des cas de SLA sont sporadiques. On soupçonne qu’un combinaison des facteurs génétiques et environnementaux sont responsables.
Familial : Aux États-Unis, la forme familiale de la maladie est observée dans une minorité de cas (5 à 10%), où les personnes héritent de la SLA d’un membre de la famille. La plupart des cas sont hérités selon un modèle autosomique dominant, ce qui signifie qu’une copie du gène modifié dans chaque cellule est suffisante pour provoquer le trouble.
Guaméen : Une forme guamanienne de la maladie a été observée dans la population de Guam où la cause est la consommation de faux palmier sagoutier, Cycas micronesia.
Physiopathologie de la SLA (partie 4 de La SLA est-elle génétique?)
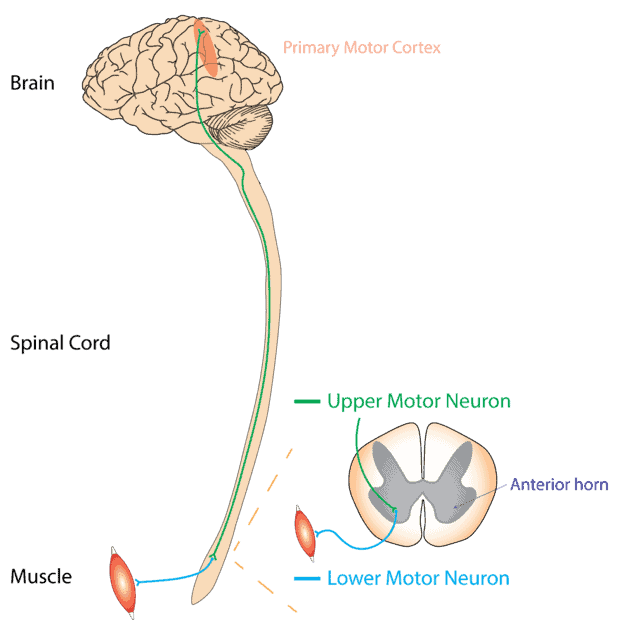
Dans la SLA, les motoneurones supérieurs et inférieurs du cerveau et de la moelle épinière sont affectés. Les motoneurones supérieurs occupent le cerveau tandis que les motoneurones inférieurs se trouvent dans la moelle épinière. Selon le site Web de ALS Pathways, il se propage généralement à partir d’un point focal, perturbant ainsi tous les motoneurones du corps et provoquant finalement la mort des motoneurones. Et de multiples facteurs sont responsables de ce type de maladie neurodégénérative. D’autres formes peuvent n’affecter que les motoneurones supérieurs (sclérose latérale primaire, PLS) ou les motoneurones inférieurs (atrophie musculaire progressive, PMA).
La dégénérescence des motoneurones supérieurs entraîne une augmentation du tonus musculaire ou une paralysie spastique. Alors que les dommages du motoneurone inférieur entraînent une augmentation de la faiblesse musculaire. Cela interfère avec le bon fonctionnement de l’activité quotidienne.
{kind=link}
Causes (Partie 5 de La SLA est-elle génétique?)
Dans la SLA, les gènes sont soupçonnés de jouer un rôle dans le risque de développer la maladie. Les études soutiennent fortement les facteurs génétiques liés à la SLA. Cependant, les études impliquant le rôle de certains des facteurs environnementaux ne sont pas très cohérentes. Dans la majorité des cas, la cause exacte n’est pas connue. Mais dans le reste de la petite cohorte (5 à 10%), des facteurs génétiques peuvent expliquer la maladie. D’autres facteurs tels que les antécédents familiaux, le sexe, la géographie, etc. peuvent également être associés à la SLA.
La SLA est-elle génétique?
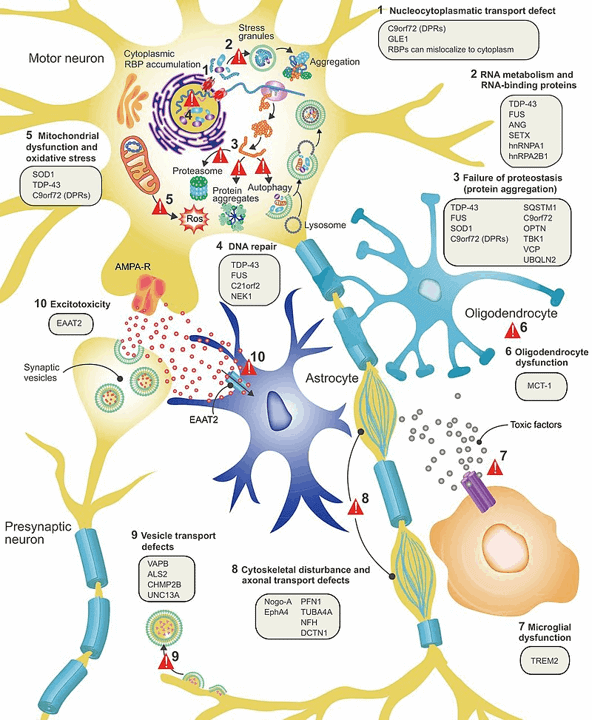
Un certain nombre de variantes génétiques sont étroitement liées à la SLA. Des mutations génétiques spécifiques associées à la maladie conduisent à une accumulation pathologique ou à une dégradation prématurée de protéines mal repliées, ce qui déclenche finalement la neurodégénérescence.
Principaux composants génétiques de la SLA
Le gène SOD1 code pour une enzyme connue sous le nom de superoxyde dismutase. Cette enzyme joue un rôle important dans le mécanisme anti-défense. Les variantes mal repliées des protéines SOD1 conduisent à une progression.
Ce gène code pour la TDP-43, une protéine qui régule l’expression génique. Le TDP-43 est essentiel aux différentes étapes du traitement de l’ARN. Certaines mutations génétiques dominantes dans le TARDBP entraînent un TDP-43 défectueux, provoquant ainsi une neurodégénérescence. Par conséquent, les changements dans la distribution et les fonctions de cette protéine ont une forte association avec la SLA.
Le gène FUS exprime la protéine FUS qui est un type de protéine de liaison à l’ARN. Certaines de ses fonctions sont similaires à celles de la protéine TDP-43. Par conséquent, la protéine FUS surveille un certain nombre de processus dans le métabolisme de l’ARN. Dans la SLA, des variantes pathogènes de ce gène provoquent parfois un repliement et une distribution inappropriés de la protéine FUS, interférant ainsi avec la physiologie cellulaire normale.
C9ORF72: Bien que le rôle exact de ce gène ne soit pas encore clair, peu de rapports ont indiqué son implication dans le transport des protéines, en particulier sous la forme d’activité endosomale et d’autophagie. Et une expression diminuée de C9ORF72 pourrait être associée à la maladie, comme indiqué dans quelques études animales. Cependant, cette régulation à la baisse à elle seule n’entraîne pas la SLA, mais fonctionne en fait dans un complexe de quelques autres protéines.
Composantes génétiques mineures de la SLA
Peu d’autres modificateurs génétiques de la SLA sont ATXN1, ATXN2, UNC13A. Les mutations de ces gènes font que les gens sensible au désordre. Mais ils ne sont pas fortement corrélés à cette maladie.
Composante non génétique de la SLA (partie 7 de La SLA est-elle génétique?)
Facteur de risque professionnel
Un nombre de rapports avoir proposé que les athlètes professionnels sont les plus susceptibles de développer la SLA. Et cela pourrait être principalement dû à de graves blessures à la tête ou à des activités physiques intenses.
D’autres exemples de exposition au travail sont des cas où des personnes sont exposées à des champs magnétiques ou à un travail physique intense. Cependant, ces études sont basées sur des associations, ne sont pas cohérentes et avec une étiologie inconnue.
Produits chimiques toxiques
Les résultats d’un étude à l’Université du Michigan à Ann Arbor (mai 2016) a confirmé la suspicion de longue date selon laquelle l’exposition à des toxines environnementales peut augmenter le risque de développer la SLA. Dans cette étude, les chercheurs ont observé une association significative entre le trouble et la présence d’hydrocarbures chlorés, de biphényles polychlorés et de biphényles polybromés.
Bêta méthylamino L-alanine (BMAA)
Le BMAA est une neurotoxine associée à la SLA. Une population autochtone vivant à Guam courrait un risque accru de contracter la maladie. Le régime alimentaire de cette population est riche en BMAA. En général, les cyanobactéries présentes dans l’eau des îles du Pacifique produisent du BMAA. Et étant donné que les habitants de Guam dépendent des fruits de mer, cela peut mettre la population à un niveau plus élevé risque .
Microbiome
Le microbiome intestinal souvent influences le résultat de la maladie, tel que rapporté dans les études animales. Microbiome intestinal études chez la souris ont démontré qu’il existe un lien important entre certaines des souches bactériennes et la SLA. De plus, dans cette étude, une souche d’Akkermansia muciniphila a tendance à ralentir la progression de la maladie chez la souris. Un autre Etude 2020 chez la souris a rapporté que les communautés microbiennes intestinales influencent le résultat de survie. Cela a été particulièrement noté, car les souris sensibles à la SLA, malgré leurs antécédents génétiques similaires, ont montré des différences significatives dans la durée de vie. Les modèles de diversité des communautés microbiennes étaient significativement différents – l’un des facteurs qui auraient pu contribuer aux différences dans la durée de survie de ces souris.
Symptômes (partie 8 de La SLA est-elle génétique?)
Les symptômes caractéristiques de la SLA sont une altération fonctionnelle du mouvement musculaire des membres supérieurs et inférieurs, des muscles bulbaires et des muscles du tronc.
Selon le Institut national des troubles neurologiques et des accidents vasculaires cérébraux , les symptômes s’accumulent progressivement. Certains symptômes précoces de la SLA comprennent:
- Fasciculations (contractions musculaires) dans le bras, la jambe, l’épaule ou la langue
- Crampes musculaires
- Muscles serrés et raides (spasticité)
- Faiblesse musculaire touchant un bras, une jambe, un cou ou un diaphragme
- Discours trouble et nasal
- Difficulté à mâcher ou à avaler
En fonction des premiers symptômes observés, la condition peut être divisée en sous-catégories. Lorsque les premiers symptômes apparaissent dans les bras ou les jambes, on parle de SLA «d’apparition des membres». Si les premières indications sont des problèmes d’élocution ou de déglutition, on parle de SLA «à début bulbaire».
La SLA progresse différemment selon les individus. Cependant, dans tous les cas, la faiblesse musculaire et l’atrophie se propagent à d’autres parties du corps jusqu’à ce que le fonctionnement devienne difficile. Les personnes aux stades avancés de la maladie finiront par ne plus pouvoir se tenir debout, marcher ou utiliser leurs membres et avaler, parler et respirer peut devenir difficile.
Dans la plupart des cas, les personnes atteintes de SLA conservent leur capacité à exécuter des processus mentaux plus élevés tels que le raisonnement, la mémoire, la compréhension et la résolution de problèmes.
La durée moyenne de survie du patient est d’environ trois à cinq ans à compter du début de la maladie, cependant, 10-20 pour cent des patients peuvent survivre plus longtemps.
Diagnostic (partie 9 de la SLA est-elle génétique?)
Surtout, les symptômes varient d’une personne à l’autre. Par conséquent, un médecin procédera à un examen approfondi en utilisant plusieurs tests . À partir de maintenant, il n’y a pas de clinique test génétique pour la SLA.
Examen physique: Dans la plupart des cas, une faiblesse musculaire croissante, une atrophie musculaire ou même des contractions musculaires conduisent les patients à consulter un médecin pour la première fois. Il est à noter que les secousses musculaires représentent à elles seules presque toujours des fasciculations bénignes. Au début, les symptômes peuvent apparaître n’importe où sur le corps. Lors de l’examen, l’apparition simultanée de signes de paralysie flasque et spastique renforce la suspicion du diagnostic de SLA.
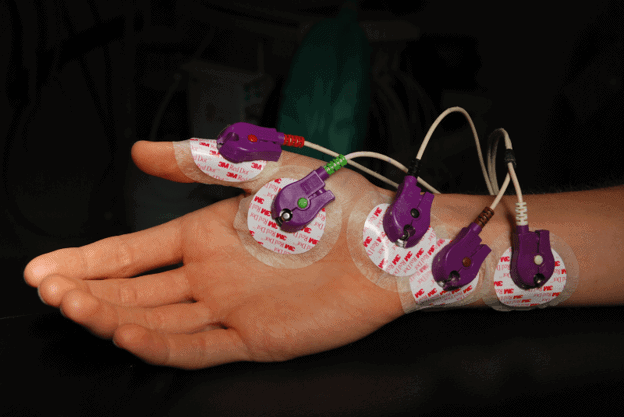
Électromyographie: Un examen électromyographique et électroneurographique est indispensable pour poser un diagnostic. L’électromyographie analyse l’activité électrique des muscles. Des critères standardisés au niveau international ont été développés pour poser le diagnostic (critères El Escorial). Selon ces critères, un diagnostic nécessite d’examiner la dégénérescence des motoneurones supérieurs et inférieurs, leur évolution, ainsi que l’absence de toute autre maladie neurodégénérative.
Robinet lombaire: Le médecin prélève un petit volume de liquide céphalo-rachidien et recherche la présence de conditions inflammatoires ou de cellules anormales. Ce test n’est important que dans les cas présentant des symptômes inhabituels.
Biopsie: Généralement, un chirurgien enlève une partie du muscle du haut du bras ou du haut de la cuisse. Le chirurgien recherche ensuite tout dommage en utilisant la coloration et la microscopie.
Traitement (partie 10 de la SLA est-elle génétique?)
Habituellement, le traitement comprend une combinaison de thérapie médicamenteuse et de thérapie orientée symptomatique. La thérapie symptomatique vise à éviter les complications de la faiblesse musculaire et à améliorer la qualité de vie du patient. Dans les modèles animaux et les cellules souches humaines, génétique études ont identifié des cibles médicamenteuses potentielles, ce qui pourrait être un pas en avant vers le développement de la pharmacothérapie.
Parfois, les gens peuvent vouloir subir des tests génétiques, en particulier dans les cas ayant des antécédents familiaux de SLA. Ensuite, dans de tels cas, le conseiller en génétique peut aider à évaluer non seulement les antécédents familiaux, mais aussi les antécédents médicaux et tout autre risque potentiel. Pour la SLA familiale, un résultat de test positif est généralement observé 60-70 pour cent des cas.
Thérapie neuroprotectrice
Le riluzole est le médicament couramment utilisé en thérapie neuroprotectrice. Ce médicament inhibe la destruction des cellules nerveuses motrices en bloquant la libération du neurotransmetteur glutamate par les neurones.
L’administration de riluzole prolongerait la survie d’environ trois mois et prolongerait les premiers stades de la SLA. Cependant, il convient de noter que la progression de la maladie ne peut être totalement stoppée par le riluzole.
Depuis 2017, la Food and Drug Administration des États-Unis a accordé l’approbation d’un autre médicament Edaravone. Il ralentit de manière indicative le développement de la SLA.
Thérapie des symptômes
Les symptômes suivants sont souvent traités avec des thérapies appropriées:
- Crampes musculaires
- La dysphagie
- Salivation
- Trouble de la parole et communication
- Rires et pleurs pathologiques
- Troubles respiratoires
- Dépression, trouble du sommeil et anxiété
Actualités récentes (2020)
Des chercheurs du Trinity College de Dublin ont lancé le Essai clinique de phase 1 de thérapie génique au Centre de recherche clinique, St.James Hospital Dublin (à compter du 1er septembre 2020). Cette thérapie génique cible le gène C9ORF72. La société pharmaceutique américaine Biogen a parrainé cet essai clinique.
As-tu aimé cet article? Assurez-vous de consulter les autres articles de la Bibliothèque de recherche sur la nébuleuse !