Table of contents
Nebula Genomics DNA Report for ALS
Is ALS genetic? We created a DNA report based on a study that attempted to answer this question. Below you can see a SAMPLE DNA report. To get your personalized DNA report, purchase our Whole Genome Sequencing!

| This information has been updated to reflect recent scientific research as of May 2021. |
What is ALS?
Amyotrophic lateral sclerosis ALS belongs to the group of motor neuron diseases and is a rare degenerative disease of the nervous system. The disease affects nerves and motor neurons. Therefore, activities such as walking, talking, chewing, or any other that requires voluntary muscles, gradually weaken.
This leads to progressive limitations in activities of daily living. Although we have no cure for ALS, doctors can slow down how fast the disease progresses.
Other names for the disease are Lou Gehrig’s syndrome or Charcot’s disease. The latter was the describer of the condition, Jean-Martin Charcot. Lou Gehrig was a New York Yankees baseball player who retired after doctors diagnosed him. Gehrig’s fate made the rare disease known to a large public for the first time.
The famous Ice Bucket Challenge from 2014 was instrumental in raising funds for research (Information source: ALS Association: Ice Bucket Challenge).

Research into the causes and potential treatments for the disease is ongoing. National organizations such as ALS Therapy Development Institute (TDI), the ALS Association, and the Centers for Disease Control and Prevention (CDC) all funded projects in this area in 2020.
Researchers at Trinity College Dublin have initiated the Phase 1 clinical trial of gene therapy at the Clinical Research Facility, St. James Hospital Dublin (as of September 1, 2020). This gene-based therapy targets the C9ORF72 gene. The U.S. pharmaceutical company Biogen has sponsored this clinical trial.
Is ALS Genetic?
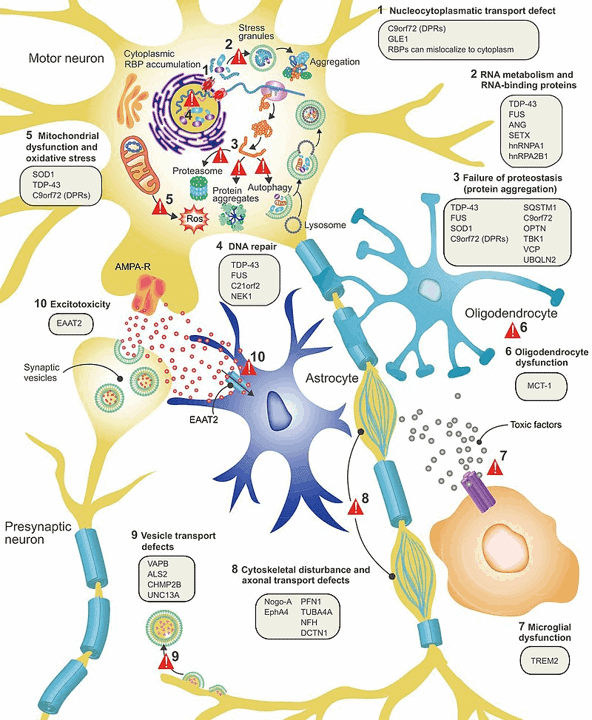
Experts have closely linked a number of genetic variations to the disorder. Specific gene mutations associated with the disease lead to the pathological accumulation or premature degradation of misfolded proteins, ultimately triggering neurodegeneration.
Major genetic components in ALS
SOD1: The SOD1 gene codes for an enzyme known as superoxide dismutase. This enzyme plays an important role in the anti-defense mechanism. The misfolded variants of SOD1 proteins lead to ALS progression.
TARDBP: This gene encodes TDP-43, a protein that regulates gene expression. TDP-43 is crucial to various stages of RNA processing. Some dominant genetic mutations in TARDBP results in faulty TDP-43, thereby causing neurodegeneration. Hence, changes in the distribution and functions of this protein have a strong association with ALS.
FUS: The FUS gene expresses the FUS protein, which is a type of RNA binding protein. Some of its functions are similar to those of the TDP-43 protein. Therefore, the FUS protein monitors several processes in RNA metabolism. Pathogenic variants of this gene sometimes cause improper folding and distribution of the FUS protein, thereby interfering with normal cellular physiology.
C9ORF72: Although the exact role of this gene is not yet clear, few reports have indicated its involvement in protein transport, especially in the form of endosomal activity and autophagy. A few animal studies have shown that a decreased expression of C9ORF72 might be associated with disease. However, this downregulation alone does not result in the disorder, but in fact, works in a complex of a few other proteins.
Minor genetic components of ALS
Experts note that ATXN1, ATXN2, and UNC13A have caused a few other familial cases. Mutations in these genes likely make people susceptible to the disorder. But they do not strongly correlate with this disease.
Current Research on ALS
In 2021, authors published a research study titled “Metal (loid)s role in the pathogenesis of amyotrophic lateral sclerosis: Environmental, epidemiological, and genetic data” in Environmental Research. It aimed to evaluate the possible role of environmental factors in the pathogenesis of the disorder.
ALS researchers conducted the study in the industrial Briga area in the province of Novara (Piedmont region, North Italy). This area is characterized by a higher incidence of sporadic ALS (sALS). Results show that patients did not carry recurrent mutations or an excess of mutations associated with sALS (SOD1, TARDBP, FUS, C9ORF72), indicating that certain environmental factors must significantly contribute to sALS. It further reiterated that the higher incidence of sALS in the Briga area may be related to environmental metal(loid)s contamination, along with other environmental factors.
According to another study titled “Value of systematic genetic screening of patients with amyotrophic lateral sclerosis” and published in the Journal of Neurology, Neurosurgery & Psychiatry, genetic screening of patients with ALS can enhance clinical care. Specifically, they focused on whether routine targeted sequencing of 44 ALS genes would have a significant impact on diagnosis and care. The study concluded that such routine screening could impact clinical care in 21% of cases.
The paper further confirms that variants within known ALS-linked genes are of potential clinical importance in 42% of patients.
Epidemiology
As of 2014, the CDC has reported 16,000 cases of ALS in the United States. An accurate record regarding its very recent statistics is not much known. In the US, about 5-7 cases exist for every population of 1,00,000. It occurs in people of all ages, but doctors usually diagnose it between 55-75 years.
However, it’s worth noting that the condition rarely affects younger patients between 25 and 35 years of age. It is also more common in men than in women (the gender ratio is about 1.5:1).
Forms
Primarily there are three main types of ALS.
Sporadic ALS: This form of ALS occurs randomly without any probable cause. A majority (90 to 95%) of the ALS cases are sporadic. Experts suspect that a combination of genetic and environmental factors is responsible.
Familial: In the U.S., doctors see the familial form of the disease in a minority of the cases (5 to 10%), where people inherit ALS from family members. Guamanian: Doctors have observed a Guamanian form of the disease in the population of Guam, where the cause is the consumption of false sago palm, Cycas micronesica.
Pathophysiology of ALS
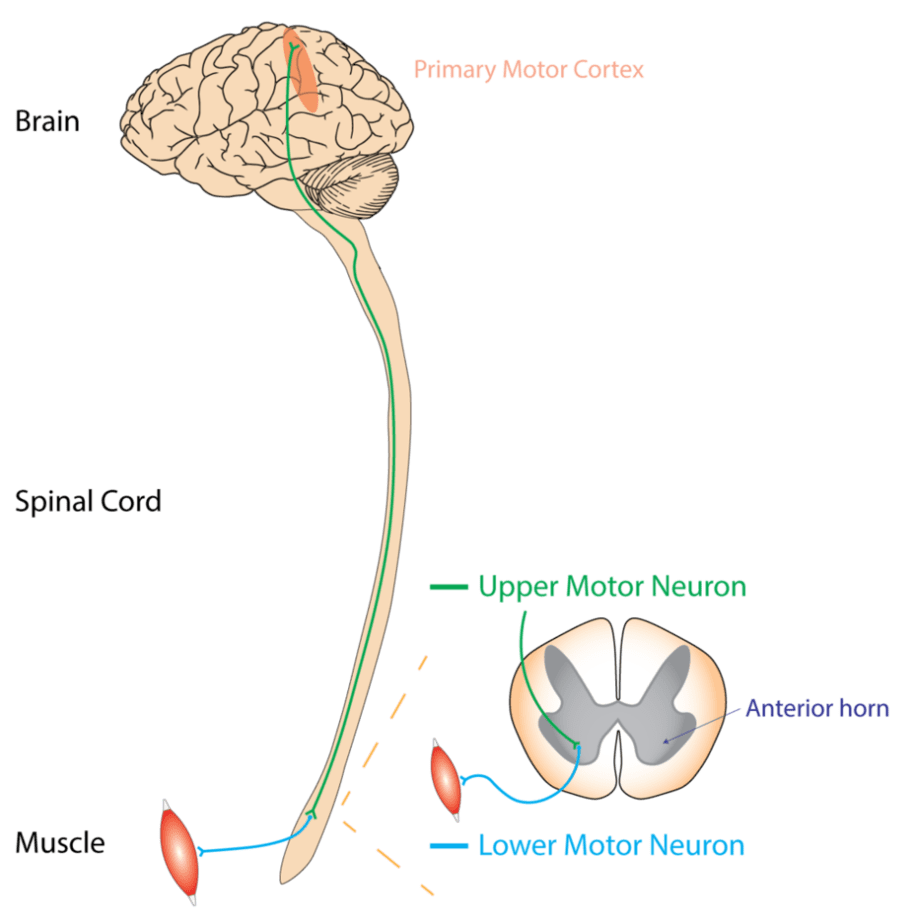
The disorder affects both the upper and lower motor neurons in the brain and spinal cord. The upper motor neurons occupy the brain, while the lower motor neurons are in the spinal cord.
According to the website of ALS Pathways, it usually spreads from a focal point, thereby disrupting all the motor neurons in the body and ultimately causing the death of motor neurons. And multiple factors are responsible for this type of neurodegenerative disease. Other forms may only affect the upper motor neurons (primary lateral sclerosis, PLS) or the lower motor neurons (progressive muscular atrophy, PMA).
The degeneration of the upper motor neurons leads to increased muscle tone or spastic paralysis while the damage of the lower motor neuron leads to an increase in muscle weakness. This interferes with the proper functioning of daily activity.
Causes
Experts suspect genes of playing a role in the risk of developing ALS. Studies strongly support genetic factors linked to ALS. However, studies implying the role of some of the environmental factors are not very consistent.
In a majority of cases, we do not know the exact cause. But in the rest of the small cohort (5-10%), genetic factors may explain the condition. Other factors such as family history, gender, geography, etc., might also have associations with ALS.
Non–genetic component of ALS
Occupational risk factor
Several reports have proposed that professional athletes are most likely to develop the condition. This could be mainly due to more instances of severe head injuries or intense physical activities, which are risk factors for ALS. Experts use this theory to link the condition with military service, especially combat veterans.
Other examples of workplace exposure are cases where people are exposed to magnetic fields or strenuous physical work. However, such studies are association-based, not consistent, and with unknown etiology.
Toxic chemicals
The results of a study at the University of Michigan in Ann Arbor (May 2016) confirmed the long-standing suspicion that exposure to environmental toxins might increase the risk of developing the disorder. In this study, the researchers observed a significant association between the condition and the presence of chlorinated hydrocarbons, polychlorinated biphenyls, and polybrominated biphenyls.
Beta methylamino L-alanine (BMAA)
BMAA is a neurotoxin associated with the disorder. An indigenous population living in Guam has reportedly been at a heightened risk of having the disease.
The diet of this population is rich in BMAA. Generally, cyanobacteria in the Pacific islands water produce BMAA. And given that the people of Guam depend on seafood, this may put the population at a higher risk of ALS.
Microbiome
The gut microbiome often influences the disease outcome, as reported in animal studies. Gut microbiome studies in mice have demonstrated an important link between some of the bacterial strains and ALS. What’s more, in this study, a strain of Akkermansia muciniphila tends to slow down the disease’s progress in mice.
Another 2020 study in mice reported that gut microbial communities influence the survival outcome. The ALS-susceptible mice, despite having similar genetic backgrounds, showed significant differences in lifespan. The diversity patterns of the microbial communities were significantly different – one of the factors that could have contributed towards the differences in the survival span of those mice.
Symptoms
The hallmark signs and symptoms of ALS are functional impairment of the muscle movement in the upper and lower extremities, the bulbar muscles, and the trunk muscles.
According to the National Institute of Neurological Disorders and Stroke, symptoms accumulate gradually. Some early symptoms of ALS include:
- Fasciculations (muscle twitches) in the tongue, arm, shoulder, or leg
- Muscle cramps
- Tight and stiff muscles (spasticity)
- Muscle weakness affecting an arm, neck, leg, or diaphragm
- Slurred and nasal speech
- Difficulty with chewing or swallowing
Depending on the first symptoms observed, doctors can further divide the condition into subcategories. When symptoms first begin in the arms or legs, they refer to it as “limb onset” ALS. If the first indications are speech or swallowing problems, they refer to it as “bulbar onset” ALS.
The condition progresses differently in different individuals. However, in all cases, muscle weakness and atrophy spread to other parts of the body until functioning becomes difficult. People with advanced stages of the disease will eventually not be able to stand, walk or use their limbs. Swallowing, speaking, and breathing may become difficult.
In most cases, people with ALS do not lose their ability to complete higher mental processes such as reasoning, understanding, remembering, and problem-solving.
The average survival time of the patient is about three to five years from the onset of the disease; however, 10-20 percent of patients living with ALS can survive for a longer duration.
Diagnosis
Mostly, the symptoms vary from one person to another. Hence, a health care professional will conduct a thorough examination using multiple tests. As of now, there is no clinical genetic testing for ALS.
Physical examination: Most of the time, patients will visit a doctor for the first time when they experience increasing muscle weakness, muscle atrophy, or even muscle twitches.
Electromyography: An electromyographic and electroneurographic examination is indispensable for making a diagnosis. Electromyography analyses the electrical activity in the muscles. Internationally standardized criteria have been developed for making the diagnosis (El Escorial criteria). According to these criteria, a diagnosis requires examining the degeneration in the upper and lower motor neurons, their progress, and the absence of any other neurodegenerative disease.
Spinal tap: The physician draws out a small volume of cerebrospinal fluid and looks for the presence of inflammatory conditions or abnormal cells. This test is important only in cases with unusual symptoms.
Biopsy: Usually, a surgeon removes a small portion of the upper arm or upper thigh muscle. The surgeon then looks for any damage using staining and microscopy.
Treatment
Usually, treatment comprises a combination of drug therapy and symptomatic-oriented therapy. Symptomatic therapy intends to help avoid complications of muscular weakness and improve the patient’s quality of life. In animal models and human stem cells, genetic studies have identified potential drug targets, which could be a step forward towards developing drug therapy.
Sometimes, people might want to undergo genetic testing, especially in cases with a family history of ALS. Then, in such cases, the genetic counselor may help in evaluating not only the family history but also the medical background and any other potential risks. For familial ALS, a positive test result is usually observed in 60-70 percent of ALS cases.
Neuroprotective therapy
Riluzole is a common drug in neuroprotective therapy. This drug inhibits the destruction of motor nerve cells by blocking the release of the neurotransmitter glutamate from neurons
The administration of riluzole reportedly prolongs survival by about three months and extends the earlier stages of ALS. However, it should be noted that the progression of the disease cannot be entirely stopped by riluzole.
As of 2017, the US Food and Drug Administration has approved yet another drug Edaravone. It indicatively slows down disease development.
Symptom therapy
The following symptoms are often treated with appropriate therapies:
- Muscle cramps
- Dysphagia
- Salivation
- Speech disorder and communication
- Pathological laughter and crying
- Respiratory failure
- Depression, sleep disorder, and anxiety
Did you like this article? Be sure to check out the other posts in the Nebula Research Library!
June 21, 2022