Relatório de DNA da Nebula Genomics para ALS
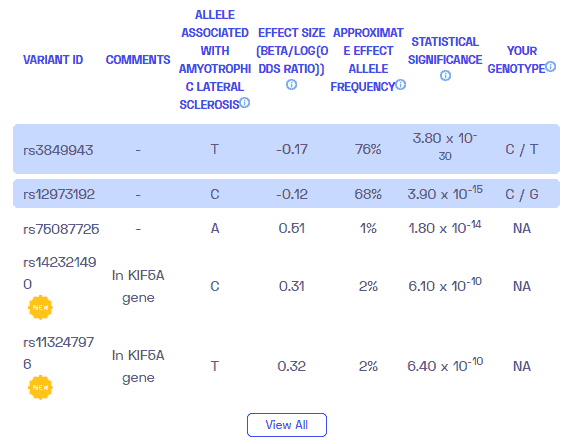
ALS é genético? Criamos um relatório de DNA com base em um estudo que tentou responder a essa pergunta. Abaixo você pode ver um relatório de amostra de DNA. Para obter seu relatório de DNA personalizado, adquira nosso Sequenciamento do genoma completo !

informação adicional
O que é ALS? (Parte 1 de O ALS é genético?)
Esclerose lateral amiotrófica (ALS) pertence ao grupo das doenças do neurônio motor e é uma doença degenerativa rara do sistema nervoso motor. Afeta adversamente os nervos e os neurônios motores. Portanto, qualquer atividade que requeira o uso de músculos voluntários, como caminhar, falar ou mastigar, enfraquece gradualmente com o tempo nesta doença. Isso leva à limitação progressiva das atividades de vida diária. Embora não haja cura para a ALS, a rapidez com que a doença progride pode ser retardada.
Outros nomes para a doença são síndrome de Lou Gehrig ou doença de Charcot. Este último foi o descritor da condição, Jean-Martin Charcot. Lou Gehrig era um jogador de beisebol do New York Yankees e teve que se aposentar após ser diagnosticado com ALS. O destino de Gehrig tornou a doença rara conhecida por um grande público pela primeira vez. O famoso Desafio do Balde de Gelo de 2014 foi fundamental para arrecadar fundos para a pesquisa de ALS (fonte de informação: Associação ALS: Desafio do balde de gelo )
.jpg){kind=link}
Pesquisas sobre as causas e tratamentos potenciais para a doença estão em andamento. Organizações nacionais como ALS Therapy Development Institute (TDI) , a Associação ALS , e as Centros para Controle e Prevenção de Doenças (CDC) todos financiaram projetos nesta área em 2020.
Epidemiologia (Parte 2 de O ALS é genético?)
A partir de 2014, o CDC relatou 16.000 casos de ALS nos Estados Unidos. Um registro exato sobre suas estatísticas muito recentes não é muito conhecido. Nos EUA, cerca de 5-7 casos existem para cada população de 1.00.000. É na faixa etária de 55 a 75 anos que a doença é finalmente diagnosticada. No entanto, é importante notar que a condição raramente afeta pacientes mais jovens entre 25 e 35 anos de idade. Também é mais comum em homens do que em mulheres (a proporção de gênero é de cerca de 1,5: 1).
Formulários (Parte 3 de ALS é genético?)
Principalmente, existem três principais formulários de ALS.
ALS esporádico : Este tipo ocorre aleatoriamente sem qualquer causa provável. A maioria (90 a 95%) dos casos de ALS são esporádicos. Suspeita-se que um combinação de fatores genéticos e ambientais são responsáveis.
Familiar : Nos EUA, a forma familiar da doença é observada em uma minoria dos casos (5 a 10%), em que as pessoas herdam ALS de um membro da família. A maioria dos casos é herdada em um padrão autossômico dominante, o que significa que uma cópia do gene alterado em cada célula é suficiente para causar o distúrbio.
Guamense : Foi observada uma forma guamã da doença na população de Guam, onde a causa é o consumo de falsa palmeira sagu, Cycas micronesia.
Fisiopatologia da ELA (Parte 4 de ELA é genética?)
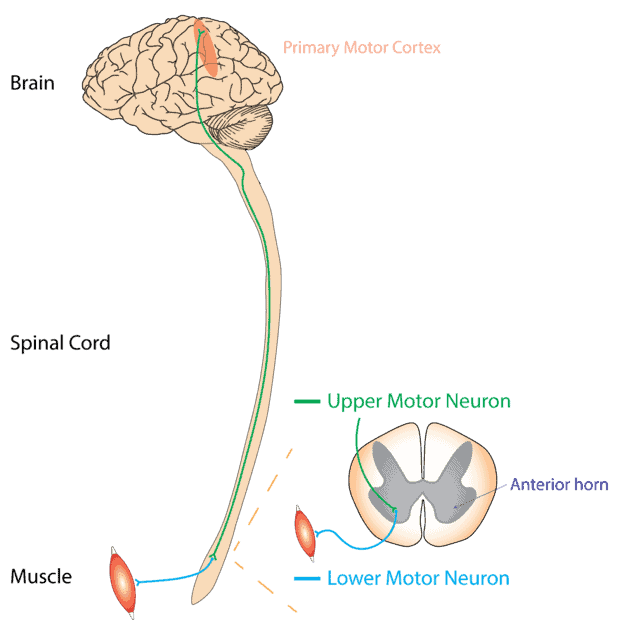
Na ELA, os neurônios motores superiores e inferiores do cérebro e da medula espinhal são afetados. Os neurônios motores superiores ocupam o cérebro, enquanto os neurônios motores inferiores estão na medula espinhal. De acordo com o site da ALS Pathways, geralmente se espalha a partir de um ponto focal, interrompendo todos os neurônios motores do corpo e, por fim, causando a morte de neurônios motores. E vários fatores são responsáveis por esse tipo de doença neurodegenerativa. Outras formas podem afetar apenas os neurônios motores superiores (esclerose lateral primária, PLS) ou os neurônios motores inferiores (atrofia muscular progressiva, PMA).
A degeneração dos neurônios motores superiores leva ao aumento do tônus muscular ou paralisia espástica. Enquanto o dano do neurônio motor inferior leva a um aumento da fraqueza muscular. Isso interfere no bom funcionamento da atividade diária.
{kind=link}
Causas (Parte 5 de ALS é genético?)
Na ALS, os genes são suspeitos de desempenhar um papel no risco de desenvolver a doença. Os estudos apóiam fortemente os fatores genéticos ligados à ELA. No entanto, estudos que sugerem o papel de alguns dos fatores ambientais não são muito consistentes. Na maioria dos casos, a causa exata não é conhecida. Mas no resto da pequena coorte (5-10%), fatores genéticos podem explicar a condição. Outros fatores como história familiar, gênero, geografia, etc. também podem ter associações com ALS.
ALS é genético?
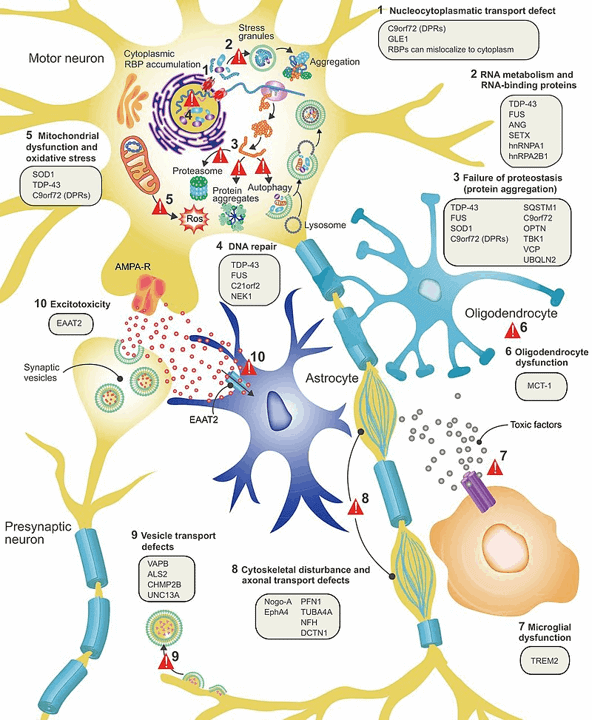
Várias variantes genéticas estão intimamente ligadas à ALS. Mutações genéticas específicas associadas à doença levam ao acúmulo patológico ou degradação prematura de proteínas mal dobradas que, em última instância, desencadeia a neurodegeneração.
Principais componentes genéticos em ALS
O gene SOD1 codifica uma enzima conhecida como superóxido dismutase. Esta enzima desempenha um papel importante no mecanismo de anti-defesa. As variantes mal dobradas das proteínas SOD1 levam à progressão.
Este gene codifica a TDP-43, uma proteína que regula a expressão gênica. O TDP-43 é crucial para vários estágios do processamento de RNA. Algumas mutações genéticas dominantes em TARDBP resultam em TDP-43 defeituoso, causando neurodegeneração. Conseqüentemente, mudanças na distribuição e funções desta proteína têm uma forte associação com ALS.
O gene FUS expressa a proteína FUS, que é um tipo de proteína de ligação ao RNA. Algumas de suas funções são semelhantes às da proteína TDP-43. Portanto, a proteína FUS monitora vários processos no metabolismo do RNA. Na ELA, variantes patogênicas desse gene às vezes causam dobramento e distribuição inadequada da proteína FUS, interferindo assim na fisiologia celular normal.
C9ORF72: Embora o papel exato desse gene ainda não esteja claro, poucos relatos indicaram seu envolvimento no transporte de proteínas, especialmente na forma de atividade endossômica e autofagia. E uma expressão diminuída de C9ORF72 pode estar associada a doenças, como observado em alguns estudos com animais. No entanto, essa regulação baixa por si só não resulta em ALS, mas, na verdade, funciona em um complexo de algumas outras proteínas.
Componentes genéticos menores de ALS
Alguns outros modificadores genéticos em ALS são ATXN1, ATXN2, UNC13A. Mutações nesses genes provavelmente fazem as pessoas suscetível para a desordem. Mas eles não se correlacionam fortemente com esta doença.
Componente não genético de ALS (Parte 7 de ALS é genético?)
Fator de risco ocupacional
Um número de relatórios ter proposto que os atletas profissionais são mais propensos a desenvolver ALS. E isso pode ser devido principalmente a ferimentos graves na cabeça ou atividades físicas intensas.
Outros exemplos de exposição no local de trabalho são casos em que as pessoas são expostas a campos magnéticos ou trabalho físico extenuante. No entanto, esses estudos são baseados em associação, não são consistentes e com etiologia desconhecida.
Químicos tóxicos
Os resultados de um estude da Universidade de Michigan em Ann Arbor (maio de 2016) confirmou a suspeita de longa data de que a exposição a toxinas ambientais pode aumentar o risco de desenvolver ALS. Neste estudo, os pesquisadores observaram uma associação significativa entre o distúrbio e a presença de hidrocarbonetos clorados, bifenilos policlorados e bifenilos polibromados.
Beta metilamino L-alanina (BMAA)
O BMAA é uma neurotoxina associada à ALS. Uma população indígena que vive em Guam corre um risco elevado de contrair a doença. A dieta dessa população é rica em BMAA. Geralmente, as cianobactérias na água das ilhas do Pacífico produzem BMAA. E dado que o povo de Guam depende de frutos do mar, isso pode colocar a população em um nível superior risco .
Microbiome
O microbioma intestinal frequentemente influências o resultado da doença, conforme relatado em estudos com animais. Microbioma intestinal estudos em ratos demonstraram que existe uma ligação importante entre algumas das cepas bacterianas e ALS. Além disso, neste estudo, uma cepa de Akkermansia muciniphila tende a retardar o progresso da doença em camundongos. Outro Estudo de 2020 em ratos relatou que as comunidades microbianas intestinais influenciam o resultado da sobrevivência. Isso foi especialmente observado, porque os camundongos suscetíveis a ALS, apesar de terem origens genéticas semelhantes, mostraram diferenças significativas na expectativa de vida. Os padrões de diversidade das comunidades microbianas eram significativamente diferentes – um dos fatores que podem ter contribuído para as diferenças no tempo de sobrevivência desses ratos.
Sintomas (Parte 8 de ALS é genético?)
Os sintomas característicos da ELA são o comprometimento funcional do movimento muscular nas extremidades superiores e inferiores, nos músculos bulbar e nos músculos do tronco.
De acordo com Instituto Nacional de Doenças Neurológicas e Derrame , os sintomas se acumulam gradualmente. Alguns dos primeiros sintomas de ALS incluem:
- Fasciculações (contrações musculares) no braço, perna, ombro ou língua
- Cãibras musculares
- Músculos tensos e rígidos (espasticidade)
- Fraqueza muscular afetando um braço, perna, pescoço ou diafragma
- Fala arrastada e nasalada
- Dificuldade em mastigar ou engolir
Dependendo dos primeiros sintomas observados, a condição pode ser subdividida em subcategorias. Quando os sintomas começam nos braços ou pernas, é conhecido como ALS de “início nos membros”. Se as primeiras indicações forem problemas de fala ou deglutição, é denominado ALS de “início bulbar”.
ALS progride de forma diferente em indivíduos diferentes. No entanto, em todos os casos, a fraqueza muscular e a atrofia se espalham para outras partes do corpo até que o funcionamento se torne difícil. Pessoas com estágios avançados da doença eventualmente não serão capazes de ficar de pé, andar ou usar seus membros e pode tornar-se difícil engolir, falar e respirar.
Na maioria dos casos, as pessoas com ALS mantêm sua capacidade de realizar processos mentais superiores, como raciocínio, memória, compreensão e solução de problemas.
O tempo médio de sobrevivência do paciente é de cerca de três a cinco anos desde o início da doença, no entanto, 10-20 por cento dos pacientes podem sobreviver por um período mais longo.
Diagnóstico (Parte 9 de ALS é genético?)
Principalmente, os sintomas variam de uma pessoa para outra. Portanto, um médico fará um exame completo usando testes múltiplos . No momento, não há clínica teste genético para ALS.
Exame físico: Na maioria dos casos, o aumento da fraqueza muscular, atrofia muscular ou até mesmo contrações musculares levam os pacientes ao médico pela primeira vez. Deve-se notar que apenas as contrações musculares quase sempre representam fasciculações benignas. No início, os sintomas podem aparecer em qualquer parte do corpo. Durante o exame, o aparecimento simultâneo de sinais de paralisia flácida e espástica reforça a suspeita do diagnóstico de ELA.
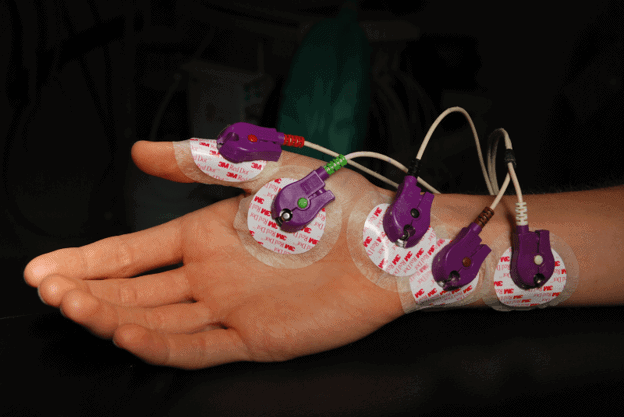
Eletromiografia: Um exame eletromiográfico e eletroneurográfico é indispensável para o diagnóstico. A eletromiografia analisa a atividade elétrica nos músculos. Critérios padronizados internacionalmente foram desenvolvidos para fazer o diagnóstico (critérios El Escorial). De acordo com esses critérios, o diagnóstico requer o exame da degeneração nos neurônios motores superiores e inferiores, sua evolução e também a ausência de qualquer outra doença neurodegenerativa.
Punção lombar: O médico retira um pequeno volume de líquido cefalorraquidiano e procura a presença de doenças inflamatórias ou células anormais. Este teste é importante apenas em casos com sintomas incomuns.
Biópsia: Geralmente, o cirurgião remove uma parte do músculo do braço ou da coxa. O cirurgião então procura qualquer dano usando coloração e microscopia.
Tratamento (Parte 10 de O ALS é genético?)
Normalmente, o tratamento compreende uma combinação de terapia medicamentosa e terapia orientada para os sintomas. A terapia sintomática visa ajudar a evitar complicações de fraqueza muscular e melhorar a qualidade de vida do paciente. Em modelos animais e células-tronco humanas, genético estudos identificaram potenciais alvos de medicamentos, o que poderia ser um passo em frente no desenvolvimento da terapia medicamentosa.
Às vezes, as pessoas podem querer se submeter a testes genéticos, especialmente em casos com histórico familiar de ELA. Então, nesses casos, o conselheiro genético pode ajudar a avaliar não apenas o histórico familiar, mas também o histórico médico e quaisquer outros riscos potenciais. Para ALS familiar, um resultado de teste positivo é geralmente observado em 60-70 por cento dos casos.
Terapia neuroprotetora
Riluzol é o medicamento comum na terapia neuroprotetora. Esta droga inibe a destruição das células nervosas motoras, bloqueando a liberação do neurotransmissor glutamato dos neurônios.
A administração de riluzol supostamente prolonga a sobrevida em cerca de três meses e prolonga os estágios iniciais da ELA. No entanto, deve-se notar que a progressão da doença não pode ser totalmente interrompida pelo riluzol.
Em 2017, a Food and Drug Administration dos EUA concedeu a aprovação de mais um medicamento, o Edaravone. Isso retarda o desenvolvimento do ALS de forma indicativa.
Terapia de sintomas
Os seguintes sintomas são frequentemente tratados com terapias apropriadas:
- Cãibras musculares
- Disfagia
- Salivação
- Distúrbio da fala e comunicação
- Riso patológico e choro
- Distúrbios respiratórios
- Depressão, distúrbios do sono e ansiedade
Notícias recentes (2020)
Pesquisadores do Trinity College Dublin iniciaram o Ensaio clínico de fase 1 de terapia genética no Clinical Research Facility, St. James Hospital Dublin (em 1 de setembro de 2020). Esta terapia baseada em genes tem como alvo o gene C9ORF72. A empresa farmacêutica americana Biogen patrocinou este ensaio clínico.
Gostou deste artigo? Certifique-se de verificar as outras postagens no Nebula Research Library !